Abstract
Risk assessment is a valuable tool for selection and optimization of rapid mycoplasma testing methods. Mycoplasma testing is a requirement at various stages in the production process for cell therapy products. Standard methods take at least 28 days and are not appropriate lot release tests for products with short shelf-lives. A review of 21 commercially available mycoplasma tests and ranking of critical risk attributes resulted in the selection of 4 methods for further evaluation. A formal risk assessment process using Failure Mode Effects Analysis (FMEA) identified areas of uncertainty in routine lot release testing. The evaluation of risk to the safety of cell therapy and tissue-engineered products should rely upon scientific knowledge and ultimately link to the protection of the patient [1].
Introduction
Mycoplasmas are a complex and unique group of bacteria belonging to the class Mollicutes and characterized by their permanent lack of a cell wall, a small genome size (0.58 – 2.2 Mbp) and a low G + C content (23 – 40 mol%) in their genomes [2]. These flexible, pleomorphic organisms can be as small as 0.2 – 0.3 μm and can achieve very high densities in cell cultures (107 – 108 organisms/ mL) without discernable changes in pH or turbidity [3]. There are currently more than 200 known species in 9 genera, many of which are pathogenic. The vast majority of cell culture contaminants belong to only 6 species of human, bovine, or porcine origin: M. hyorhinis, M. arginini, M. salivarium, M. orale, M. fermentans, and A. laidlawii.
Routes of mycoplasma contamination in a cell therapy manufacturing process include materials, personnel, donor tissue, and cross-contamination. Although manufacturing in a cGMP environment ensures control of these sources of contamination, lot release testing is still necessary to ensure product safety. Standard culture methods take at least 28 days to complete and are therefore not timely lot release tests for cell therapy products having abbreviated shelf-lives as short as 72 hours. One such product, with a 6-day shelf-life, is an autologous chondrocyte implant product to repair articular cartilage damage in the knee. Figure 1 gives an overview of the process.
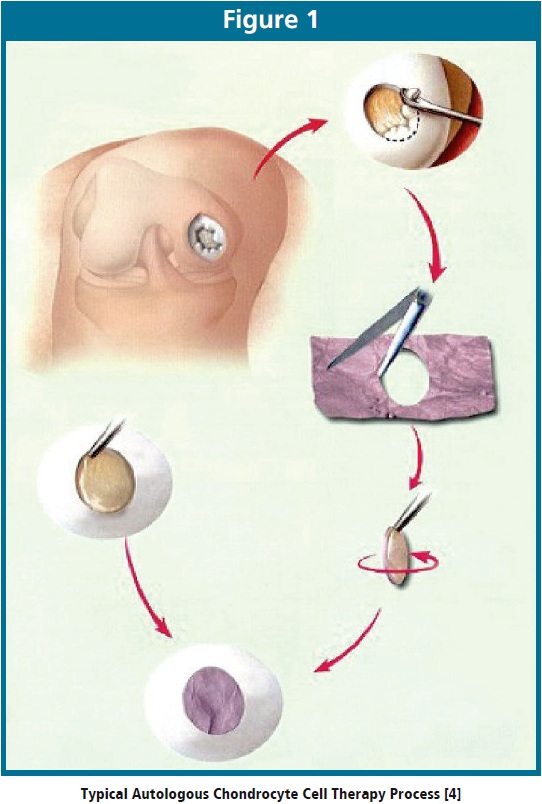
First, a surgeon obtains a small cartilage biopsy. Technicians in a cGMP production facility culture the cells over a period of several weeks to increase the cell number from a few hundred thousand to several million prior to seeding onto a collagen membrane. Once the culturing process is complete, the clinical site receives the product for implantation. During the implantation procedure, the surgeon makes an incision in the knee and implants the cell therapy product into the defect [5].
In recent years, both commercial manufacturers and academic research laboratories have developed a number of rapid mycoplasma test methods with the potential to address the lot release timing for products with short shelf-lives. These tests use molecular biology techniques, PCR most commonly, as well as biochemical or immunological reactions to generate and detect a response specific to mycoplasma. Biochemical techniques include enzyme assay, ELISA, and cell-surface receptor detection. Immunological techniques detect specific antibody-antigen interactions and molecular techniques other than PCR include DNA-binding with fluorescence detection. In contrast to standard direct culture methods that take at least 28 days to complete, most of these indirect tests can take less than 24 hours to complete. Table 1 provides a summary of potential mycoplasma assay technologies for consideration.
Methodology
Review of commercially available methodology is a valuable first step when developing a new rapid microbiological test. Ranking critical risk attributes such as testing time, specificity, and sensitivity allows selection of methods for further evaluation at a cell therapy testing facility. Critical risk attributes are elements of the test design or process that could ultimately impact patient treatment if not mitigated. Table 2 summarizes a rating scale useful for a preliminary risk assessment of rapid mycoplasma tests.
A rating of 0 correlates to an unacceptable parameter value, 1 correlates to a parameter value that may be acceptable, and 2 correlates to an optimal parameter value. The total score for each kit is the product of the ratings; the tests with the highest scores warrant on-site evaluation.
The on-site evaluation should minimally consider kit specificity and sensitivity. To evaluate specificity, analysis of spiked and unspiked samples provides an assessment of sample interference with the kits. In addition, based on recommendations in the European Pharmacopoeia chapter on Mycoplasmas [6], analysis of bacterial species with a close phylogenetic relationship to mycoplasma (Streptococcus, Clostridium, Lactobacillus) provides an assessment of likely bacterial cross-reactivity with the kits. To evaluate sensitivity, analysis of serial dilutions of purified mycoplasma DNA provides a measure of the limit of detection. In addition, data about other kit characteristics such as stability, ease of use, and logistics issues may be important when making a final selection.
Finally, a formal risk assessment process using Failure Mode and Effects Analysis (FMEA) identifies areas of uncertainty in designing a rapid mycoplasma test based on a commercial method and in performing routine lot release testing using this method prior to implementation into a cGMP system. FMEA is a method of reliability analysis intended to identify failures that may have significant consequences to the performance of the design or process considered. A “failure mode” describes the way that a design or process could fail. The FMEA process works as follows:
• Divide the test design or test process into items or steps, respectively.
• Identify and examine possible failure modes to determine possible causes, effects, and current controls.
• Rate each item or step for Frequency, Detectability, and Severity.
• Calculate a Risk Priority Number (RPN) = Frequency x Detectability x Severity.
• Compare the RPN to a pre-established table based on pre-defined rules to determine whether mitigation is required.
As a prerequisite, each organization needs to determine the values in the RPN table and mitigation strategies based on the nature of the business and its tolerance to risk.
Results and Discussion
In 2007, there were 21 mycoplasma tests on the market, some with the potential to meet the lot release time constraints inherent to autologous cell therapy products. A preliminary risk analysis resulted in the selection of 4 commercial methods for further evaluation in a cell therapy production facility.
Several issues identified during detailed discussions with the manufacturer’s technical experts eliminated one of the methods. The remaining 3 methods employed real-time PCR techniques, evaluated at the test lab for specificity, sensitivity, and other metrics. Table 3 summarizes the results of the qPCR kit evaluation testing.
Specificity
None of the kits produced false positive results when tested with un-spiked samples. Kits 2 and 3 produced false negative results when challenged with samples spiked with positive control DNA. However, these results were most likely due to poor DNA recovery during extraction or inhibition from inadequate sample cleanup. It is noteworthy that similarly spiked samples with poor DNA recovery produced positive results using Kit 1.
Kit 3 only claimed to detect 8 species, while the other 2 kits claimed to have primers designed to detect the majority of species. Kit 3 was not tested for cross-reactivity because it had previously been eliminated due to unacceptable kit stability. Kit 2 reacted when challenged with S. pyogenes but did not react when challenged with C. sporogenes or L. acidophilus. Kit 1 did not react when challenged with any of the 3 bacterial species tested.
Sensitivity
It was difficult to conduct a meaningful comparison of sensitivity for the following reasons. First, the lowest amount of DNA tested with Kit 3 was 50 copies per reaction because unacceptable kit stability eliminated it prior to testing additional dilutions. Next, the manufacturer of Kit 2 could not provide quantitative information about the positive control. Even though comparing the kits is difficult, it is noteworthy that Kit 1 detected single-digit numbers of DNA copies per reaction.
Other Metrics
Among the other kit characteristics compared, Kit 3 included a sample preparation from which <5% DNA recovery was obtained, Kit 2 did not include sample preparation, while the manufacturer of Kit 1 developed an optimized sample preparation and customized it for specific cell therapy applications. In addition, Kit 1 is relatively easy to use, especially compared with Kit 2, which is too overly complex for routine quality control testing. Kit 3 has not been optimized for use on all widely-available commercial instrumentation, while the other 2 kits both provided specific thermocycler programs for each instrument. Finally, delivery and logistics issues experienced with Kit 2 were extreme, making implementation into routine operations unfeasible.
As a result, elimination of 2 more methods (Kits 2 and 3) based on insufficient specificity, poor reagent stability, inadequate sample preparation, or unnecessary complexity left the 4th method (Kit 1) for further optimization, validation, and implementation.
FMEA
Prior to optimizing, validating, and implementing Kit 1, a Failure Mode and Effects Analysis identified elements in the design and steps in the process contributing to the highest risk of failure. Use of commercially available risk assessment software facilitated the FMEA process. The Design FMEA found areas of risk primarily associated with sample configuration and sample preparation, proposing 14 mitigation activities. These activities included choosing an optimal sample configuration, optimizing the sample preparation for recovery, and qualifying the kit vendor. The Process FMEA found areas of risk associated with routine analysis using a qPCR lot release test, proposing 15 mitigation activities. These activities included developing clear standard operating procedures to address reagent preparation, sample handling, and verification of instrument settings, providing molecular biology training, and obtaining feedback from regulatory agencies on the validation plan.
Visualizing the results of the FMEA using Pareto analysis allows comparison of the relative risk associated with design or process components. Pareto analysis is a statistical technique used to select the design or process components contributing to the majority of the risk. For example, Figure 2 shows that the Design FMEA found the most risk in selection of sample configuration while the Process FMEA found the most risk in developing clear procedures.
Conclusions
Application of a risk-based approach to development, validation, and implementation of rapid microbiological testing systems is consistent with the objectives of ICH Guideline Q9 [7]. Using this approach, a rapid mycoplasma test kit based on real-time PCR technology was optimal for validation and implementation as a lot release test for cell therapy products. In summary, the level of effort, formality, and documentation should be commensurate with the level of risk, while the evaluation of risk to the safety of cell therapy and tissue-engineered products should rely upon scientific knowledge and ultimately link to the protection of the patient.
References
1. Duguid J, Kielpinski G, du Moulin GC, Seymour B. Application of a Risk-Based Approach to Optimize a Rapid Mycoplasma Test for Cell Therapy and Tissue-Engineered Products. Atlanta, GA: AAPS Annual Meeting and Exposition; 2008.
2. Bacterial Pathogens. Mycosafe Diagnostics GmbH Web site. 2008. Available at: http://www.mycosafe.at/mycosafe/ mycosafe.nsf/alldocs/7C21DDE60B3859B3C125700E00576B02? OpenDocument. Accessed August 5, 2008.
3. FAQ. Bionique® Testing Laboratories, Inc. Web site. 2008. Available at: http://www.bionique.com/. Accessed August 5, 2008.
4. ACI-MaixTM. Matricel GmbH Web site. 2008. Available at: http://www.matricel.de/assets/images/Verigen_MACI.jpg. Accessed August 5, 2008.
5. MACI® Implant – Advanced Cartilage Repair. Genzyme Corporation Web site. 2008. Available at: http://www.maci. com/pat/detail/mc_pat_advanced_cartilage_repair.asp. Accessed August 5, 2008.
6. 2.6.7. Mycoplasmas. In: European Pharmacopoeia. 6th ed. Strasbourg, FR: European Directorate for the Quality of Medicines; 2007.
7. Q9. Quality Risk Management. ICH Harmonised Tripartite Guideline. Current Step 4 version. November 9, 2005.
John Duguid is a scientist at Genzyme responsible for developing and implementing rapid microbiological assays. Previously, he managed QC cell therapy operations. Mr. Duguid received his BS in Chemistry from the University of Michigan. Prior to Genzyme, he worked as an analytical chemist at Abbott Laboratories and Arthur D. Little.
Barbara Seymour is Director of Process Development for cell therapies at Genzyme Biosurgery. Barbara has worked on autologous, allogenic, xenogeneic, and combination cellular therapy products for over 20 years including multiple U.S. and EU clinical trials. She holds a BS and MBA degree.
Grace Kielpinski is a project manager at Genzyme responsible for technology development. In this role, Ms. Kielpinski pursues new technologies for potential use in cell therapies and provides a bridge from research and development to operations. Ms. Kielpinski has over 20 years of operations experience including Quality Control, manufacturing, and materials management.
Gary C. du Moulin, Ph.D., M.P.H. is Senior Director of Quality Compliance for Genzyme Biosurgery where he oversees the development and execution of robust quality systems for cell therapy and tissue engineered products. Dr. du Moulin joined Genzyme in 1995 after working for six years developing quality systems for cellular therapies for the treatment of renal cell carcinoma. Prior to his industrial experience, he spent 15 years in the Department of Anaesthesia at Harvard Medical School (Beth Israel Hospital) in Boston attaining the rank of Assistant Professor of Anesthesiology. He has more than 150 publications in the areas of microbiology, epidemiology, and the regulation and quality control of living cells as a therapeutic modality.
Dr. du Moulin received his B.S. in 1969 from the Military College of Vermont-Norwich University, an M.S. degree from Northeastern University, and M.P.H. and Ph. D. degrees from Boston University. Dr. du Moulin currently serves on the Gene Therapy, Cell Therapy, and Tissue Engineering Expert Committee of the United States Pharmacopoeia and chairs the ad hoc advisory panel for fetal bovine serum. He serves on the editorial board of Regenerative Medicine and is RAC certified and past Chairman of the Editorial Board of the Regulatory Affairs Professionals Society Magazine, RAPS Focus. He is retired from the U.S. Army Reserve at the rank of Colonel after 38 years of service.
To contact the author please, email him directly at: [email protected]