Introduction
As the pharmaceutical industry has changed, so have the ways scientists use dissolution testing. In today’s drug development process, scientists must carefully balance the need for at least three different types of dissolution workflows. The workflows are listed below in sequential order from early to late stages of drug development.
1. Low volume (250mL) bio-relevant dissolutions [1,2] for formulation selection.
2. Dissolution media selection for method development of a selected formulation.
3. Routine quality control (QC) dissolution testing to support formulation stability, process scale-up and clinical release studies.
Although the volume and dissolution media for these dissolution workflows may differ, dissolution tests occupy a significant amount of analyst time. Dissolution tests are labor intensive because they often require the analyst to perform many steps manually. The analyst must fill the dissolution pots with specific media, obtain samples from the dissolution pot, filter or centrifuge the samples, clean the dissolution apparatus, and finally place the samples onto an HPLC for analysis. While the time-consuming nature of dissolution testing has sparked the development of several automated dissolution testing systems, vendors have focused on automating the third type of workflow mentioned above [3]. Although these automated dissolution systems reduce the amount of dedicated analyst time, they are not capable of conducting bio-relevant dissolutions or efficient automated dissolution media selection for method development [4].
Additionally, with the increasing importance of bio-relevant dissolution testing as a tool to guide formulation development [5], automated dissolution systems need to be capable of performing more than just QC dissolution methods. In essence, the automated dissolution systems scientists use must also change. The ideal automated dissolution system would be capable of automating:
• Low volume (250mL) bio-relevant dissolutions.
• Dissolution media screening experiments for method development.
• Routine QC dissolution tests to support stability, scale-up and clinical release studies.
• The physical movement of dissolution samples from the dissolution bath to an HPLC and its acquisition of samples.
To address gaps in current automated dissolution workflows, Merck scientists have developed automated workflows using a commercially available automated dissolution system. These workflows are capable of meeting the needs of both early and late stage development scientists. Examples of these automated workflows will be shown for each type of dissolution workflow.
Biorelevant Dissolution Studies
In the pharmaceutical development environment, leveraging science to reduce costs and maintain high quality deliverables is ideal. A key area of cost reduction can be associated with clinical and preclinical testing. In the early stages of developing a pharmaceutical formulation, reducing the number of formulations taken into animal studies can achieve significant savings. A reduction in the number of animal studies can be achieved by using bio-relevant dissolution experiments to provide a formulation rank order [5].
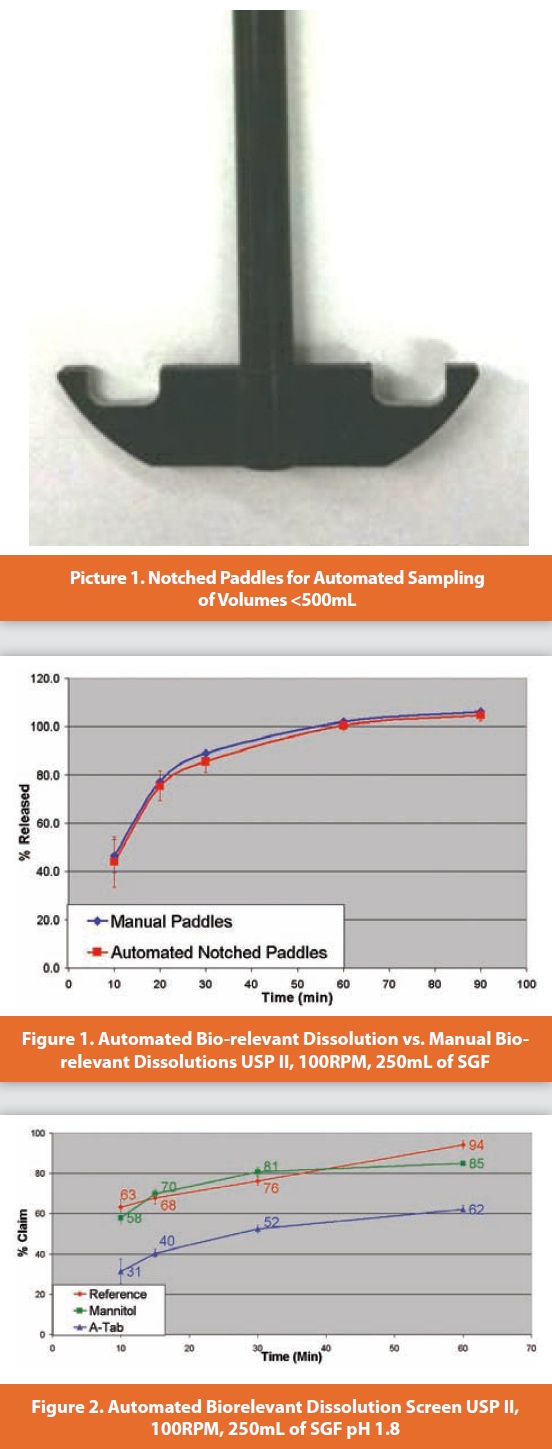
Bio-relevant dissolution testing uses media which mimic the fluids found in the digestive tract [1,2,5]. Bio-relevant dissolution can drive early formulation selection and optimize the application of invivo studies. However, bio-relevant testing also requires significant analyst effort because sampling usually occurs manually due to the low volumes (<500mL) being employed. Most typical autosamplers cannot collect samples at a 250mL volume because sampling cannula becomes entangled with the spinning paddle. While it may be possible to increase the dissolution volume to 500mL, this consumes twice the quantity of API and may not be feasible given the often limited availability of API in the early development environment. For the same sampling related reasons, most current automated dissolution systems cannot sample volumes <500mL and consequently cannot conduct bio-relevant dissolution experiments.
Using specially designed notched paddles (See Picture 1), automated sampling can occur down to volumes below 250mL. Consequently, fully automated bio-relevant dissolution testing can be achieved using this system. The notched paddles are easily interchangeable and can quickly be removed and replaced with traditional normal USP II paddles for QC testing.
Figure 1 compares automated and manual 250mL bio-relevant SGF dissolutions conducted on the same formulation. The dissolution profiles from the automated system with the notched paddles and manual dissolution with traditional paddles were comparable.
Using this automated dissolution setup, an example of automated, low volume, bio-relevant dissolution was performed on a BCS class II Active Pharmaceutical Ingredient (API). This particular API is an acidic salt of a weak base, has a strong pH dependent solubility profile and was formulated in two different formulation matrices.
The automated 250mL SGF biorelevant dissolutions used the USP II apparatus. Samples were acquired at 10, 15, 30, and 60 minutes, clarified with 0.45um polytetrafluoroethylene filters and stored into a capped 96 sample well plate. After the automated dissolution completed, the well plate was robotically transferred to the integrated HPLC and an automated HPLC run sequence was created. A two minute isocratic HPLC method was used to quantitate the samples. The results from these formulations were compared against a reference formulation and can be seen in Figure 2.
The automated bio-relevant dissolution predicted that formulation 2 would be inferior to the reference and formulation 1 supplies. Subsequent in-vivo testing showed the same relationships between the formulations as the automated dissolution results. These biorelevant dissolution results provided extremely valuable information and demonstrated the importance of both pH and excipient selection for this compound. The results from these experiments also allowed for the optimal formulation candidate to be appropriately prioritized for in-vivo studies.
Automated Dissolution Method Development
In early phase drug development (i.e. pre-clinical candidate selection to Phase I), some knowledge of an API’s solubility is available. The behavior of the API in a solid dosage form is unknown. Therefore, it is critical to select appropriate dissolution method conditions for the formulation as these conditions may remain with the product throughout its drug development lifetime. One such important consideration for the dissolution method is the evaluation of different media compositions.
Dissolution media screening experiments typically focus on differences in pH, surfactant type and/or surfactant levels. Traditional automated dissolution systems have a single, large weigh vessel for media dispensing. This design allows all dissolution pots to be filled with only one media type per run. Therefore, the typical automated dissolution system cannot efficiently screen media conditions in a single automated dissolution run.
The automated dissolution system used has individual vessels capable of dispensing different media into each of the 8 pots. With the ability to select from 4 different media source lines, duplicate preparations of up to 4 different media types can be run in a single automated dissolution experiment.
The case study below highlights the benefits of using automation for method development purposes. This API salt has a strong pH dependent solubility profile within the pH range of the human gastric region. It was critical to select a media pH that is discriminating and capable of obtaining full release.
The results shown in Figure 3 were generated in a single automated dissolution run using a USP II apparatus, with a 900mL fill volume and 75RPM paddle speed. Samples were taken at 5, 10, 15, 30, 45, 60 and 90 minutes and were clarified using a 0.45um polytetrafluoroethylene filter. After the 60 minute timepoint, the paddle speed was increased to 200RPM to ensure full release was obtained at the 90 minute timepoint. Samples were analyzed using the fully integrated HPLC which employed a 5 minute HPLC isocratic method.

Data from the single, automated dissolution method development experiment was generated with minimal analyst time. Four different pH conditions were evaluated with two replicates at each pH. The results from this study showed that the pH 2.0 media was the only media that generated both an acceptable dissolution profile and full release by 45 minutes. This method was successfully implemented for future formulation stability testing. Similar automated dissolution method development studies have been performed evaluating both dissolution surfactant levels and types and have shown that these too can be screened in an automated workflow.
Fully Automated Dissolution Testing
As formulations progress beyond Phase I, scale-up studies and stability studies become increasingly large. QC dissolution methods are often required to test for differences in formulation performance and / or stability changes. As the demand for dissolution testing increases, the analyst can be faced with performing a significantly higher volume of dissolution studies. Performing these tasks manually is not viable in today’s fast paced pharmaceutical development paradigm. This stage in development is often thought to be the most appropriate time to implement automated dissolution testing. The integration of an HPLC instrument, capable of running any separation condition, into an automated dissolution system would provide a significant throughput advantage. In some cases, traditional automated dissolution systems do have the ability to incorporate the HPLC analysis into the workflow. However, these systems require that the acquisition of the HPLC data is completed before the next dissolution sample is obtained. Since sampling can occur every 5 minutes, the HPLC acquisition time would need to be less than one minute per injection. This traditional approach to HPLC integration severely limits the type of HPLC method.
In the automated dissolution workflow presented below, multiple tablets from 16 different formulation scale activities were analyzed in a single fully automated dissolution queue. This was able to be accomplished because the system has the ability to accommodate up to 8 capped well plates in a sample repository hotel (see Picture 2). The well plates are automatically transferred from the hotel to the dissolution system, and then to an HPLC before finally returning to the hotel for sample retention.
Dissolution samples were acquired at 10, 30, 45 and 60 minutes and clarified using a 0.45um polytetrafluoroethylene filter. The samples were analyzed using a 2 minute HPLC run time with the integrated HPLC. Results from the study can be seen in Figure 4. From the formulation studies in Figure 4. one clear trend was observed. The wet granulation formulations (solid lines) consistently outperformed the roller compacted formulations (dotted lines). No chemical or processing liabilities were associated with the wet granulation process; therefore, it was selected as the lead process for future formulation development.
Due to the fully automated dissolution system, all 16 formulation results were available within 24 hours of sample receipt. Had these studies been conducted using the traditional manual dissolution approach, approximately three full days of dedicated analyst time would have been required to perform the manual dissolutions.
Conclusions
Automated dissolution has historically had the most significant impact in late stage pharmaceutical development. However, with the creation of new automated workflows, automated dissolution can also be successfully implemented into core, early phase drug development activities. These core development activities include, bio-relevant dissolution studies that provide formulation rank order, automated media screening for methods development, a fully automated system capable of analyzing formulations and stability samples within a QC environment.
Implementing the workflows previously described can provide significant advantages in both bio-relevant formulation knowledge and analyst time savings. These automated dissolution workflows can be easily implemented in either research or manufacturing stages and the analyst time savings can be re-directed to other valuable tasks.
References
1. Vertzoni, M.; Dressman, J. B.; Butler, J.; Hempenstall, J.; and Reppas, C. “Simulation of fasting gastric conditions and its importance for the in vivo dissolution of lipophilic compounds”, European Journal of Pharmaceutics and Biopharmaceutics, 2005, 60 (3), 413-417
2. Kalantzi, L.; Goumas, K.; Kalioras, V.; Abrahamsson, B; Dressman, J. B. and Reppas, C. “Characterization of the Human Upper Gastrointestinal Contents Under Conditions Simulating Bioavailability/Bioequivalence Studies”, Pharm. Res., 2006, 23 (1), 165-176.
3. Kozlowski, B.; Wuelfi ng, W. “Automation of Dissolution – An Evaluation of Existing Technologies and Approach to Implementation”, Am. Pharm. Rev., 2007 (May/Jun).
4. Kozlowski, B.; John, C. W. “Specialty Automation Groups and Implementation of Automated Dissolution in Early Development Settings”, Am. Pharm. Rev., 2008 (Mar/Apr).
5. Wang, Q.; Fotaki, N.; and Mao, Y.; “Biorelevant Dissolution: Methodology and Application in Drug Development”, Dissolution Technologies, 2009, 16 (3), 6-13.
Author Biographies
Christopher T. John is a Sr. Research Chemist at Merck & Co., with 10 years experience. Chris current leads the activities of a specialty analytical group in the application of automation and spectroscopic technologies. These methods have played key roles throughout formulation development. Chris received his B.S. degree in from Muhlenberg College and is pursuing his M.S. degree from Lehigh University.
Christina Bacci is a chemist at Merck & Co., in the Analytical Sciences division at West Point, PA. She has a bachelor’s degree in chemistry from Ursinus College in Collegeville, PA and a M.S. degree in chemistry from the University of California, Irvine. Christina uses dissolution to study solid dosage forms during formulation development and develops and validates analytical methods to study the quality and stability of drug products.