Introduction
The use of solid dispersions, defined broadly as involving the dispersion of a drug in a polymeric carrier via a liquid intermediate, has been a highly studied approach for drug delivery since the 1960s [1-4]. The greatest single driver for the use of this technique has been the remarkable improvements in dissolution rate that may be observed for poorly water soluble drugs, with increases that went up to orders of magnitude being frequently reported [5,6]. Development of formulation strategies for poorly water-soluble are perhaps even more important now as significant numbers of new chemical entities which show highly promising pharmacological effects for many severe diseases such as HIV and cancer, have low aqueous solubility and fall into Class II or IV of the Biopharmaceutics Classification System [7].
One particularly promising manufacturing process for the production of solid dispersions is hot melt extrusion (HME). HME has been used for polymer processing for decades and has been recently adopted for pharmaceutical use for the preparation of solid dispersion formulations [8]. It combines melting, highly intimate mixing of the drug and polymer via extrusion of their fluid mix under pressure and cooling into a continuous single step process with no involvement of solvents. The resultant mixture may be ground or pelletized and used in a range of dosage forms. Amongst the many advantages and possibilities afforded by HME is the opportunity to formulate poorly water soluble drugs in a molecularly dispersed form within an amorphous hydrophilic polymeric carrier [8,9]; this may be referred to as an amorphous solid dispersion. It is recognized that amorphous dispersions can improve dissolution as a result of the lack of a crystalline matrix alongside other potential effects such as improved wettability and the absence of drug particle aggregation. The amorphous dispersion field presents an exciting research opportunity for formulation scientists in that aspects of technology development, biorelevant dissolution studies, physical chemistry and polymer science are all incorporated. However, the physical stability of amorphous solid dispersions remains one of the most difficult issues associated with this formulation technology. Physical instability often appears in the form of phase separation in which the drug and polymer migrate and form distinct phases [10- 14] and this is believed to have a significant negative impact on the dissolution enhancement of the formulation [15]. The detection and/ or prediction of phase separation is extremely important during early formulation development as it may indicate long term instability requiring reformulation to produce an acceptable product.
In this article, we discuss the combined use of global and localized characterization methods to detect separated phases, in particular focusing on the initial stages of phase separation. Some newly developed AFM based localized characterization methods are highlighted as they have demonstrated the ability to detect low quantities of phase separation with micron to submicron dimensions.
Phase Separation – A Kinetic Process with a Thermodynamic Cause
Phase separation can be induced by external physical stress [10-14] or the supersaturation state of the drug in the polymer (in which the drug loading is significantly higher than the solid solubility of the drug in the polymer). The fundamental cause of phase separation irrespective of the stimulus is the thermodynamic instability of the formulation. A phase separated systems has, in its simplest form, domains of the two pure components. The concept arises from comparisons with binary polymeric systems, whereby phase separated system is well recognized by the presence of two distinct glass transitions corresponding to the individual components as opposed to a single intermediate transition. Phase separation in drug dispersions is more complex as the drug could be either crystalline, amorphous, and even in intermediate stages such as drug rich domains and systems containing both amorphous and crystalline drug (Fig 1). However, despite the final form of phase separated drug in the formulation, the separation is a kinetic process. This means that the separation starts with low quantity and small dimensions of the separate phases at the early stage of the separation and possible transformations of intermediate forms of separate phases during the course of separation. This often makes the situation for pharmaceutical solid dispersions even more complicated compared to binary polymeric systems and the characterization and data interpretation more challenging. For example, if a drug has certain degree of miscibility with the polymer, there may be a point at which a separate drug phase forms, with some of the drug remaining molecularly dispersed while crystallites also form via an amorphous intermediate, hence several different drug physical states may coexist simultaneously through the separation process. There is also the possibility (as yet not demonstrated but a possibility nonetheless) that the system may phase separate into conjugate systems of both components (drug rich and polymer rich domains), i.e. instead of phase separation into drug and polymer the system separates into two distinct drug-polymer mixes.

Hot melt extruded amorphous dispersions often carry the risk of phase separation during storage under ambient conditions. We have suggested that this is because the high pressure and shear forces accompanied by heating that are applied to the extruded materials during the HME process provides highly intimate mixing of the polymer and drug; however the same process also has high potential of forming a supersaturated drug-polymer system [16,17]. It is well known that the solubility of a drug in a solution changes with temperature and pressure. The same principle almost certainly applies to drug-polymer systems, with the solubility of a drug in the polymer being elevated during the extrusion process under high temperatures and pressures. However, on storage at ambient temperature and pressure, the initial molecularly dispersed system can start to separate in to different phases because of the lower solubility of the drug in the polymer at these lower temperatures and pressures. This phase separation may occur over hours or years; nevertheless it may put the formulation at risk of altered in vivo performance over the shelf life of the product and associated regulatory difficulties. Therefore the detection of phase separation in the initial stages of development and the understanding of the mechanisms and kinetics of the separation is extremely important to the formulation process.
Identification of Phase Separations
Global vs. Localized Characterization Methods
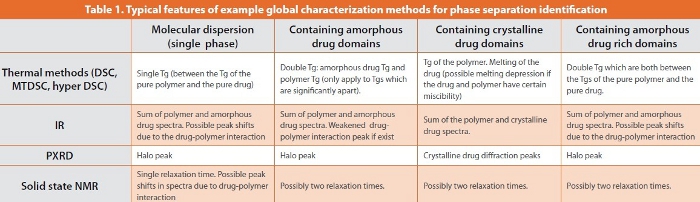
The term global characterization methods refer, in this context, to analytical techniques which test the sample as a whole, typically in quantities of a few mg. The resulting data are therefore representative of the totality of properties of the entire sample. Localized characterization methods, in contrast, measure the properties of the selected local regions of a sample and can be used to map the distribution of different components or physical states of the same substance in a sample. In terms of identifying phase separation, both types of approach have advantages at different stages of the process. At the latter stages of phase separation, when the domains sizes are relatively large (micron range), the phase separation may be easily detected using conventional global characterization methods if the quantity of the separate phase is above the limit of detection of those methods. There are many global characterization methods available which are used routinely for formulation screening including thermal analysis (DSC, MTDSC, hyper DSC), spectroscopic methods (FTIR, Raman, Terahertz spectroscopy, and solid state NMR), and PXRD (Table 1). In addition, new technological and methodological developments of the conventional global characterization methods can strengthen the identification capacity of the method. For example, thermal analysis such as DSC and MTDSC are routinely used methods for quick identification of phase separation. If a formulation contains phase separated crystalline drug, the melting of the crystalline drug should be observed in the experiment. However, depending on the miscibility of the drug and the polymer, the melting of the crystalline drug is often absent due to the dissolution of the drug in the softened glassy polymer during heating if the drug quantity is small. This may lead to incorrect interpretation as the absence of a discernible melting peak may be considered to be evidence of molecular drug dispersion, whereas it is merely a facet of slow dissolution of the drug in the molten polymer during heating [18,19]. With fast heating rate DSC, it is possible to detect drug melting by not allowing sufficient time for the dissolution process to occur (bearing in mind that melting is not rate dependent whereas dissolution is). In effect the use of fast heating rates renders the timescale of the experiment short in comparison to the time required for the sample to dissolve into the molten polymer, hence the drug melting may be observed more reliably. Recently a heat capacity measurement method using a conventional MTDSC experiment has been shown to be effective for overcoming the dissolution effect of low quality of crystalline drug in polymer during heating [16]. As seen in Figure 2, by measuring heat capacity changes during a heating and cooling cycle, it is possible to estimate the quantity of crystalline drug which is not observable through observation of the melting behavior in a conventional DSC measurement. However, there are drawbacks of global characterization methods mainly associated with the sample preparation, low specificity of the analysis and the limit of detection of the techniques. For example, obtaining accurate PXRD results requires grinding of the original solid dosage forms to produce a powder. After pulverization, the initial physical form of the drug and excipients may be changed and the results based on the powdered sample can be misleading.
For the reasons associated with HME discussed earlier, many freshly prepared HME samples are often likely to show the features associated with amorphous dispersions such as exhibiting a single Tg; however this does not by any means indicate that the sample will be stable on storage. Identification of any phase separation at its initial stage and the prediction of phase separation kinetics is at the heart of controlling the quality of HME drug products. During phase separation, the dimensions, quantity and the physical nature of the separated phases will change over time. Initially the separate phases are often low in number and of sub-micron size. This makes the detection of the phase separation difficult as it may approach or be below the detection limits of many global characterization tools. Although it may be argued that the small size and low quantity phase separation may not have profound effect on the dissolution of the formulation, if the phase separation is not identified at an early stage, further growth and crystallization of the drug may potentially cause a significant reduction in dissolution and absorption of the drug. Moreover, the distribution of the phase separation may be an important issue in some particular cases where there is significant drug concentration variation across the formulation. This may affect dissolution and be an important consideration for monitoring batch variation. For this issue, localized characterization methods can provide important complementary information. Additionally, HME can often be coupled with injection moulding to produce extrudates with any desired shape and size. Many of the extrudates are used in the final formulation as their original geometric shapes, such as implants [20]. The particular requirements of being characterized in their original form often make many global methods impossible to provide accurate analysis.
Imaging techniques combined with chemical identification is a growing stream of new characterization techniques. These include IR microscopy and imaging, MRI imaging, confocal Raman microscopy and AFM based techniques [23,24,26-28]]. However, these techniques all have different spatial resolutions. Among these, Raman imaging and AFM have the potential of reach submicron resolution [21-28]. These novel approaches can detect phase separation which are not identified by conventional analytical methods [16,17]. As seen in Figure 3, conventional DSC showed no clear presence of crystalline drug in the hot melt extruded formulations because of the absence of crystalline drug melting [17].

However, pulsed force mode AFM (PFM-AFM), which is an intermediate-contact mode AFM, revealed crystalline-like drug particles with 1-3micron diameter on the surface of the extrudates. The PFM-AFM images are obtained from measurements based on sample properties such stiff ness, adhesion, viscosity energy dissipation and sample-tip contact time. This gives the method the capacity of not only imaging at submicron scale but also to identify phases based on the tip-sample physical interactions. However, conventional AFM based techniques have a much lower capability for chemical identification compared with spectroscopic imaging techniques such as IR and Raman imaging [26-28]. For example, phase imaging conducted using tapping mode AFM and PFM-AFM can only provide phase identification based on the measurement of the physical properties of the separated phases; this in turn requires further supplementary characterization for confirmation on the actual chemical composition of the phases (either being drug or polymer or mix). Some new developments of functionalized AFM strengthen the physical property based phase identification and render chemical phase identification possible. For example, recently developed local-thermal analysis (LTA) and photothermal-FTIR (PT-FTIR) microspectroscopy can provide localized characterization of a sample at micron to submicron scale with no additional sample preparation [16,17,29,30]. These methods are both derivatives of AFM whereby the tip is replaced by a miniaturized heater. In the case of LTA, the probe position is measured as a function of tip temperature, hence allowing the identification of phase transitions such as melting via the measurement of probe penetration into the sample, while PT-FTIR allows the operator to obtain IR spectra on highly specific regions of a sample via measurement of the heat fluctuations as a function of frequency when an IR beam is focused on the sample. Localized thermal analysis can therefore be used to detect the difference in the thermal properties of different phases in the solid dispersion [27-29], while PT-FTIR microspectroscopy can provide a photothermal spectrum of the chemical makeup of the AFM probe landing area [30,31].
These approaches are particularly useful for HME samples in terms of examining not only the micron to submicron size domains but also their distribution. Both the surface and the centre of the extrudate can be first imaged using tapping mode AFM or PFM-AFM [28], hence some indication of phase separation can be suggested by the methods based on the samples physical properties. Once the image is obtained, chemical identification of phase separations can be carried out using local-thermal analysis and/or PT-FTIR microspectroscopy. For example, as seen in Figure 4, the surface of the examined extrudates showed uniform single Tg indicating the amorphous dispersion of the drug (paracetamol) in the polymer [28]. However, at the inner core of the extrudates (cross-sectional face), the LTA results showed great variation and many of the tested points revealed double tip penetration. These relate to the initial softening of the material when Tg is approached, followed by melting of the crystalline paracetamol [28]. However, it was possible to obtain information on the presence of low quantity crystalline paracetamol and to detect significant variation on drug distribution across the extrudates using the localized techniques which was not possible using global characterization methods such as DSC, IR, and PXRD. PT-FTIR can provide spectroscopic information of the local regions of the sample which can further assist the analysis of the drug distribution, providing information on the local drug concentration and the physical state of the drug (as amorphous or crystalline) [17,30]. As seen in Figure 5, the PT-FTIR microspectroscopy spectra correspond to different sampling points on the surface and the cross section of the hot melt extruded formulations. Little variation is observed in the spectra of the selected local regions on the surface and cross-section of the unsaturated formulation (with low drug loading), whereas clear differences are present in the spectra of the selected local regions on the surface and the cross-section of the formulation with high drug loading [17].
Conclusions
Global and localized characterizations have their advantages and disadvantages in phase separation identification for solid dispersions. As a kinetic process, the quantity, physical nature (amorphous or crystalline) and the chemical makeup of the separate phases change with time. Although phase separation can occur with solid dispersions prepared by other methods such as fusion and solvent evaporation, those prepared by HME are more vulnerable to phase separation at ambient conditions over aging because of the intensive pressure and shear often accompanied with a relatively high temperature during the process. If the separate phases are relatively abundant, global characterization methods are ideal and rapid identification approaches. However at early time points phase separation may present in low quantities and small dimensions and may be missed by global characterization methods because of their limits. In these cases localized characterization can be very useful. Therefore, the combined use of global and local analysis at different stage of phase separation may be the key to ensure a clear and accurate understanding of phase separation process in HME solid dispersion formulations.
References
- K. Sekiguchi and N. Obi, Studies on absorption of eutectic mixtures: I. A comparison of the behavior of eutectic mixtures of sulfathiazole and that of ordinary sulfathiazole in man Chem. Pharm. Bull. 9 (1961) 866–872
- W.L. Chiou and S. Riegelman, Pharmaceutical applications of solid dispersions, J. Pharm. Sci. 60 (1971) 1281–1303
- J.L. Ford, The solid dispersions, Pharm Acta Helv. 61 (1986) 69–88
- D.Q.M. Craig, The mechanism of drug release from solid dispersion in water-soluble polymers, Int. J. Pharm. 231 (2002) 131-144
- A.T.M. Serajuddin , Solid dispersions of poorly water-soluble drugs: early promises, subsequent problems and recent breakthroughs. J. Pharm. Sci. 88 (1999) 1058–1066
- C. Leuner, J. Dressman, Improving drug solubility for oral delivery using solid dispersion, Euro J. Pharm. Biopharm. 50 (2000) 47-60
- C.J.H. Porter, C.W. Pouton, J.F. Cuine, W.N. Charman, Enhancing intestinal drug solubilisation using lipidbased delivery systems, Adv. Drug. Del. Rev. 60 (2008) 673-691
- M.M. Crowley, F. Zhang, M.A. Repka, S. Thumma, S.B. Upadhye, S. K. Battu,.J.W. McGinity, C. Martin Pharmaceutical applications of hot-melt extrusion: Part I, Drug Dev. Ind. Pharm. 33 (2007) 909-926
- J. Breitenbach, Melt extrusion: from process to drug delivery technology, Euro. J. Pharm. Biopharm. 54 (2002) 107-117
- .J. Marsac, A.C.F. Rumondor, D. E. Nivens, U. S. Kestur, L. Stanciu, L.S. Taylor, Effect of Temperature and Moisture on the Miscibility of Amorphous Dispersions of Felodipine and Poly(vinyl pyrrolidone), J. Pharm. Sci. 99 (2010) 169-185
- J.Baird, R. Olayo-Valles, C. Rinaldi, L.S. Taylor, Effect of Molecular Weight, Temperature, and Additives on the Moisture Sorption Properties of Polyethylene Glycol, J. Pharm. Sci. 99 (2010) 154-168
- A.C.F. Rumondor, L.A. Stanford, L.S. Taylor, Effect of Polymer Type and Storage Relative Humidity on the Kinetics of Felodipine Crystallization from Amorphous Solid Dispersions. Pharm. Res. 26 (2009) 2599-2606.
- A.C.F. Rumondor, I. Ivanisevic, S. Bates, D.E. Alonzo, L.S. Taylor, Evaluation of Drug-Polymer Miscibility in Amorphous Solid Dispersion Systems. Pharm. Res. 26 (2009) 2523-2534.
- A.C.F. Rumondor, P.J. Marsac, L.A. Stanford, L.S. Taylor, Phase Behavior of Poly(vinylpyrrolidone) Containing Amorphous Solid Dispersions in the Presence of Moisture. Molecular Pharmaceutics. 6 (2009) 1492-1505.
- S. Janssens, C. Roberts, E.F. Smith, G. Van den Mooter, Physical stability of ternary solid dispersions of itraconazole in polyethyleneglycol 6000/hydroxypropylmethylcellulose 2910 E5 blends , Int J Pharm 355 (2008) 100-107
- S. Qi, P. Belton, K. Nollenberger, N. Clayden, M. Reading, D. Q.M. Craig, Characterisation and prediction of phase separation in hot melt extruded solid dispersions: a thermal, microscopic and NMR relaxometry study, Pharm. Res. 27 (2010) 1869-1883
- S. Qi, P. Belton, K. Nollenberger, A. Gryczke, D. Q.M. Craig, Characterisation of phase separation in hot melt extruded solid polymeric dispersions containing a poorly water-soluble drug: a combined AFM, spectroscopic and localised thermal analysis approach, (2010) Analy. Chem. submitted
- G.R. Lloyd, D.Q.M. Craig and A. Smith , A calorimetric investigation into the interaction between paracetamol and polyethylene glycol 4000 in physical mixes and solid dispersions. Eur. J. Pharm. Biopharm. 48 (1999) 59–65
- G.R. Lloyd, D.Q.M. Craig and A. Smith , An investigation into the melting behaviour of binary mixes and solid dispersions of paracetamol and PEG 4000. J. Pharm. Sci. 86 (1997) 991–996
- T. Quinten, T.R.M. De Beer, C. Vervaet, J. P. Remon, Evaluation of injection moulding as a pharmaceutical technology to produce matrix tablets. Eur. J. Pharm. Biopharm. 71 (2009) 145-154
- K.L.A. Chan, N. Elkhider, S.G. Kazarian, Spectroscopic Imaging of Compacted Pharmaceutical Tablets, Chem. Eng. Res. Des. 83 (2005) 1303-1310
- L. Maurer, H. Leuenberger, Terahertz pulsed imaging and near infrared imaging to monitor the coating process of pharmaceutical tablets, Int. J. Pharm. 370 (2009) 8-16
- Multivariate data analysis for Raman imaging of a model pharmaceutical tablet, Anal. Chim. Acta, 545 (2005) 262-278
- B. Vajna, I. Farkas, A. Szabó, Z. Zsigmond, G. Marosi, Raman microscopic evaluation of technology dependent structural diff erences in tablets containing imipramine model drug, J. Pharm. Biomedical. Analy. 51 (2010) 30-38
- T. Tajiri, S. Morita, R. Sakamoto, M. Suzuki, S. Yamanashi, Y. Ozaki, S. Kitamura Release mechanisms of acetaminophen from polyethylene oxide/polyethylene glycol matrix tablets utilizing magnetic resonance imaging, Int. J Pharm. 395 (2010) 147-153
- I. Weuts, F. Van Dycke, J. Voorspoels, S. De Cort, S. Stokbroekx, R. Leemans, M. E. Brewster, D. Xu, B. Segmuller, Y.A. Turner, C. J. Roberts, M. C. Davies, S. Qi, D.Q.M. Craig, M. Reading, Physicochemical properties of the amorphous drug, cast fi lms and spray dried powders to predict formulation probability of success for solid dispersions: Etravirine, J. Pharm. Sci. 99 (2010) 196-208
- J. Zhang, M. Bunker, X. Chen, A. P. Parker, N. Patel, C.J. Roberts Nanoscale thermal analysis of pharmaceutical solid dispersions, Int. J. Pharm. 380 (2009) 170-173
- S. Qi, A. Gryczke, P. Belton, D.Q.M. Craig, Characterisation of solid dispersions containing paracetamol prepared by hot melt extrusion using thermal, microthermal and spectroscopic analysis, Int. J. Pharm. 354 (2008) 158-167
- L. Harding, S. Qi, G. Hill, M. Reading, D.Q.M. Craig, The Development of Microthermal Analysis and Photothermal Microspectroscopy as Novel Approaches to Drug-Excipient Compatibility Studies, Int. J. Pharm. 354(2008) 149-157
- X. Dai, J. G. Moff at, A.G. Mayes, M. Reading, D.Q. M. Craig, P.S. Belton, D.B. Grandy, Thermal Probe Based Analytical Microscopy: Thermal Analysis and Photothermal Fourier-Transform Infrared Microspectroscopy Together with Thermally Assisted Nanosampling Coupled with Capillary Electrophoresis, Analy. Chem. 81(2009) 6612-6619
- J.G. Moff at, A.G. Mayes, P.S. Belton, D.Q. M. Craig, M. Reading, Compositional Analysis of Metal Chelating Materials Using Near-Field Photothermal Fourier Transform Infrared Microspectroscopy, Analy. Chem. 82(2010) 91-97
Author Biographies
Duncan Craig holds the Chair in Pharmaceutics and is Head of the School of Pharmacy, University of East Anglia. His research involves the physical characterisation of dosage forms in relation to performance using a range of thermal, rheological and imaging approaches.
Sheng Qi is a lecturer in Pharmaceutics at the School of Pharmacy, University of East Anglia. Her research mainly involves the development of lipid based dosage forms and the use of novel physicochemical characterisation approaches for solid dosage forms.