The opinions expressed in this publication do not necessarily reflect the on-going evolving philosophy of the research organizations represented.
Introduction
The Biopharmaceutics Classification System [1,2] (BCS) is not only a useful tool for obtaining waivers for in-vivo bioequivalence studies but also for decision making in the discovery and early development of new drugs. It is because BCS is based on a scientific framework to classify drugs using solubility and permeability as the rate limiting steps in oral absorption. In general, the four necessary steps for a drug to be absorbed are 1) release of drug from dosage forms, 2) maintenance of dissolved state throughout gastrointestinal (GI) track, 3) permeation of drug molecules through GI membrane into hepatic circulation, 4) enterohepatic metabolism that influences the systemic availability. The Biopharmaceutical Drug Disposition Classification System (BDDCS) proposed by Y. Wu and L Z Benet [3] completes the absorption process by including the fourth rate-limiting step of first pass metabolism.
The evaluation of oral absorption is critical to the discovery of orally potent compounds. Consequently the determination of solubility, permeability, and metabolic stability have been fully integrated by most pharmaceutical companies as an integral part of high throughput screening (HTS) and lead optimization. A multitude of automated 96 well systems to screen solubility, permeability and metabolism are commercially available. These oral absorption screening tests are often referred as pharmaceutical profiling [4] which data can be used for preliminary BCS classification.
It is not unusual that polymorphism issues have delayed either toxicological or clinical programs throughout the industry. With the shortened development cycle time, some companies have recognized the need to move polymorph screening into early development preferably prior to animal studies. Early selection of salt/polymorph enables a consistent pharmacokinetic exposure from one study to another right from the beginning so that the development cycle may be compressed. In the past, the polymorph screening was not done until after POC for two reasons: 1) Conserve resources considering the high attrition rates in early development. 2) Regulatory authorities consider different polymorphic forms as having pharmaceutical sameness but not for different salt form. A new salt qualifies a new NDA filing (505b2 route) while a new polymorph can be filed via ANDA route. It is because different polymorphs may give same bioavailability if dissolution is not a rate limiting step such as BCS class 1 compounds with high solubility and high permeability. Whereas the presence of different counter ions in the salt forms may impart varied safety and efficacy profiles. As a result, the salt form is usually selected prior to initiation of drug development program.
There is plenty of incentive for the pharmaceutical industry to move polymorph screening into early development. In the past decades, pharmaceutical manufacturers have experienced disrupted supply or recalled commercial products when polymorph changes occurred in the manufacture of API or during drug product storage [5]. It is because polymorphic forms differ in their crystal lattice energy that results in different solubility and therefore dissolution rates and eventually bioavailability. The late discovery of rotonavir thermodynamic stable form with a lower solubility and oral bioavailability led to a 6 month disruption in drug supply, loss of hundreds of millions of dollars in sales, and suboptimal therapy for thousands of patients [6]. Moreover, the cautionary tale of ranitidine litigation on its original anhydrate form teach the industry not only that late discovery of new polymorphs can delay the progression of clinical trials but also that late disclosure of polymorphs may translate into less profit. Once the compound is known, there is no stopping others to discover a more bioavailable, stable and convenient polymorph than the original form. It is why nowadays, a company will start patent proceedings for discovered polymorphs as soon as the publication of the initial chemistry patent.
The objective of this article is to describe a BCS based polymorph/salt form and formulation strategies that can often lead into a minimum design for higher efficiency and lower cost. The BCS based drug discovery strategy has largely been in place in the design of lead compounds with a set of solubility, permeability and metabolic properties. This information can very well be used in the prioritization of salt/polymorph screening in early pharmaceutical development. The goal is to shorten the development cycle and streamline clinical studies without delay due to polymorph/salt issues. This approach has been able to support the R&D Productivity Model which has been put in place since 2002as described by B. Ruffolo [7]. The Productivity Model calls for delivery of 12 IND candidates per year, for which ~18-22 new molecular entities are evaluated for salt/polymorph. The overall resources required are roughly 0.5 full time equivalents over 8-12 weeks per compound. The BCS based salt/polymorph selection strategy is in line with FDA’s year 2000 risk management and 2004 critical path initiatives.
BCS Distribution of Pharmaceutical Compounds
With the advent of high throughput screening around 1990, a shift of lead compound biopharmaceutical characteristics into less drug-like has propelled discovery departments in pharmaceutical companies to utilize computational chemistry to optimize solubility permeability, and metabolic stability such as Lipinski’s “rule of 5” [8]. However, solubility of new pipeline compounds seems not improved but moving toward a higher degree of hydrophobicity in the quest of nanomolar binding efficiency (IC50) and high biological potency (ED50). As a result, the new drug pipeline tends to have lower solubility resulting in an increase of BCS 2 compounds from ~30% to ~60%. In 2004, Dressman et al [9] assigned BCS classification to the 130 compounds listed by WHO on the Model list of Essential Medicines. In 2005, C. Y. Wu and L. Z. Benet expanded the list to include 141 marketed compounds2. The distribution of these 141 old compounds in each of the BCS class 1, 2, 3 and 4 are 39%, 31%, 23% and 7% for marketed product. Comparatively, the distribution of over 100 new compounds from Wyeth in each of the BCS class 1, 2, 3 and 4 are 15%, 57%, 12% and 23% as reported by M. S. Ku in 2006 [10]. Similarly, the distribution of GSK new compounds in each of the BCS class 1, 2, 3 and 4 are 19.9%, 62.6%, 8.4% and 9.2% as reported by J. Baldoni in the 2007 AAPS/FDAWorkshop of BE, BCS and Beyond. Other companies such as Pfizer [11] and Bristol Meyer Squibb [12] reported similar problem with poor solubility for lead compounds.
Salt Formation: An Enabling Technology to Improve Solubility and Stability
The use of salt forms to improve solubility and bioavailability is well documented and reviewed. Bighley et al. provided a systematic compilation of various drug salts and their effects on PK and bioavailability [13]. However, maximizing the bioavailability potential via salt formation needs to be considered early in the development cycle since regulatory agency recognize salt as a different compound from the free base or free acid. Salt Formation can not only increase bioavailability but also improve solid state stability whenever the stability of the free base or free acid is in question. Because the salt formation involves proton transfer with a change in electronic density distribution so the reactivity of the nucleophilic or electrophilic functional groups are modified. The author throughout the years was able to stabilize many IND lead compounds via salt formation. Since 70% of pipeline compounds are ionic in nature, approximately ~10% of which offers opportunity for stabilization in the author’s experience. The exemplary mechanism of stabilization are prevention of decarboxylation of carboxylic acid, hydrolysis of sulfonamide, and hydroxamic acids, dimer and adduct formation by nucleophilic amines and oxidation of free amine groups. For more detailed discussions on the impact of solid state properties on stability, refer to the two recent excellent reviews by Yoshioka and Stella [14] and Guillory and Poust [15].
Polymorphism Regulatory Guidance Linked with BCS Classification
FDA recognizes the importance of polymorphism. Upon issuance of BCS guidance in 2000, FDA immediately issued a draft polymorphism guidance highlighting the utility of BCS in determining whether a polymorph specification is required for the drug substance and/or drug product. In the final guidance issued in July 2007 [16], FDA states, “For a drug whose absorption is only limited by its dissolution, large differences in the apparent solubility’s of the various polymorphic forms are likely to affect BA/ BE. On the other hand, for a drug whose absorption is only limited by its intestinal permeability, differences in the apparent solubility’s of the various polymorphic forms are less likely to affect BA/BE.” FDA further states “Furthermore, when the apparent solubility’s of the polymorphic forms are sufficiently high and drug dissolution is rapid in relation to gastric emptying, differences in the solubility’s of the polymorphic forms are unlikely to affect BA/BE.” In other words, FDA is saying that polymorphism is not critical for BCS class 1 and 3 compounds but is for BCS class 2 and 4 compounds. The FDA guidance has applied BCS nicely in the establishment of polymorph specification.
The decision trees in FDA’s guidance stipulate for the drug substance, if there are known polymorphs with different apparent solubility’s and not all polymorphs are highly soluble as defined by BCS criteria, a polymorph specification is necessary. The guidance further stipulate for the drug product, if there is sufficient concern that a polymorph specification in the drug product be established or if the drug product performance testing (e.g., dissolution testing) does not provide adequate controls when the polymorph ratio changes, a polymorph specification is necessary. FDA recommends considering only those polymorphs that are likely to form during manufacture of the drug substance, manufacture of the drug product, or while the drug substance or drug product is in storage. The guidance clarifies that there may not be a concern if the most thermodynamically stable polymorphic form is used or the same form is used in a previously approved product of the same dosage form. Finally, FDA states that drug product performance testing (e.g., dissolution testing) can generally provide adequate control of polymorph ratio changes for poorly soluble drugs, which may influence drug product BA/BE. It is clear that FDA expects only in rare cases would polymorphic form characterization in the drug product be necessary. It can be concluded that the polymorph control is for the purpose of controlling oral bioavailability and that for highly soluble and highly permeable compounds, polymorphism is of little concern.
Similar decision trees are in the ICH Q6A Guidance [17] including the following four questions: 1) Can different polymorph be formed? 2) Do the forms have different properties? (Solubility, stability, melting point), 3) Does drug product performance testing provide adequate control if polymorph ratio changes (e. g. dissolution), and 4)? Is drug product safety, performance, or efficacy affected? The bottom line is for BCS class 1drug with high solubility and high permeability; polymorphism is unlikely to impact drug product performance and therefore specification is unnecessary.
Ambiguity in the Definition of Polymorphism
Both ICH and FDA decide, for convenience, to encompass a broader range of solid forms in the polymorphism guidance stating “Polymorphic forms in the context of this guidance refer to crystalline and amorphous forms as well as solvate and hydrate forms”. Scientifically speaking, hydrates are a subclass of solvates with the solvent being water, both of which have different chemical composition from the neat form and at best can be referred to as psudopolymorphs. Cocrystals although not mentioned in the regulatory guidance, have gained popularity recently due to its enhanced solubility characteristics. Cocrystals can be formed between two chemical molecules via mostly H-bonding whereas the salt is via proton transfer forming ionic bonds. Solvates can be viewed as a subclass of cocrystal where the other component is a solvent. Cocrystals can contain either stoichiometric or non-stoichiometric amounts of the other molecules such as solvent, water, carboxylic and amino acids or sugars like sorbitol. An example of nonstoichiometric cocrystals is the pseudohydrate with water molecules tunneling in and out of the crystal lattice without changing the powder x-ray diffraction (pXRD) pattern. Another example of non-stoichiometric solid forms is what the author calls “Soft Salt” with incomplete formation of salt form upon API manufacturing where pXRD conforms but the counterion assay fall short of the stoichiometric amount. 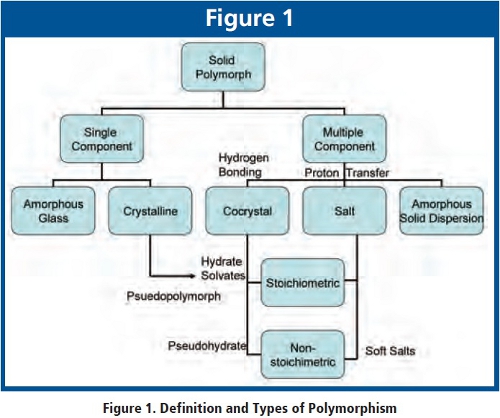 Another characteristic of Soft Salts is the easy exchange of counterions when in contact with excipients. In 2002, the author discovered that a HCL salt of a primary amine drug exchanged the counterion with Na starch glycolate resulting in an insoluble glycolate salt with slowed dissolution. Since then several incidences of salt exchanges were noted between active and excipients such as magnesium stearate, sodium lauryl sulfate, and sodium croscarmellose. Similar experience has been reported by a Merck author [18] in the April 2007 IQPC conference. Dr. Kwong positively identified using near IR the slow down of dissolution for a Merck compound is due to salt exchange. As to amorphous forms, the guidance states “Amorphous forms consist of disordered arrangements of molecules that do not possess a distinguishable crystal lattice.” The disordered arrangement by definition can vary in endless ways. Perhaps the better descriptor is “non-crystalline forms”. The hierarchy of solid form definitions are summarized and described in Figure 1.
Another characteristic of Soft Salts is the easy exchange of counterions when in contact with excipients. In 2002, the author discovered that a HCL salt of a primary amine drug exchanged the counterion with Na starch glycolate resulting in an insoluble glycolate salt with slowed dissolution. Since then several incidences of salt exchanges were noted between active and excipients such as magnesium stearate, sodium lauryl sulfate, and sodium croscarmellose. Similar experience has been reported by a Merck author [18] in the April 2007 IQPC conference. Dr. Kwong positively identified using near IR the slow down of dissolution for a Merck compound is due to salt exchange. As to amorphous forms, the guidance states “Amorphous forms consist of disordered arrangements of molecules that do not possess a distinguishable crystal lattice.” The disordered arrangement by definition can vary in endless ways. Perhaps the better descriptor is “non-crystalline forms”. The hierarchy of solid form definitions are summarized and described in Figure 1.
Limitation in Intellectual Property Value Enhancement
In recent years, the pharmaceutical industry has increasingly focused its attention on salt/polymorph discovery due to the realization that a new salt/polymorph can translate into more or less profit. A salt/polymorph exhibiting unique advantage is a patentable composition of matter which affords additional protection to the family of intellectual properties for a new drug. However, as stated by W. C. McCrone back in 1965 “The number of forms known for a given compound is proportional to the time and money spent in research on that compound.” There is no guarantee that a competitor won’t discover a new salt/polymorph of your drug that is better than the patented ones. It is because the chemistry of crystallization is far from ideal and the formation of polymorphs depends on heterogeneous surface chemistry and time-dependent kinetic phenomenon. The recent finding by Matzger [19] that growing crystals on different polymeric surfaces produced different polymorphs of the same compound, is hardly a surprise. Nevertheless, with hundreds of millions of dollars at stake, a company that develops a new drug will patent all discovered salts/polymorphs preferably prior to the publication of the first chemistry patent on the new compound. At this early stage of the development phase, it is not possible and necessary to discover all the possible crystal forms. Pharmaceutical companies have options to first find a developable salt/polymorph form and then expand the screening at a later stage for more rare occurring polymorphs to strengthen the intellectual property position for the product.
Screening Methodology for Thermodynamic Stable Polymorph
The key objective of polymorph screening is to identify the thermodynamically stable form in the target formulation environment during manufacture and storage. Even for the first animal pharmacokinetic study using an aqueous suspension for oral gavage, hydrate formation in the suspension can result in a significant change in solubility, uniformity and suspend-ability, which would give non-reproducible data. It is why polymorphism needs to be evaluated ahead of animal studies. Several high throughout methods with automated or semi-automated sample handling and characterization for salt and crystal form screening has recently been reported [20, 21]. While these new methodologies provide extra capacities for solid form discovery, it does not replace detailed form characterization to understand interrelationship of various forms.
To obtain various solid forms, a dozen solvents and anti-solvents utilizing four different crystallization methods (evaporative, cooling, gas diffusion and anti-solvent addition) are investigated at 100 mg per solvent. The samples are examined under polarized microscope for birefringence. The crystalline samples are subject to differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) to determine whether it is a mixture or a pure polymorph. The pure polymorphs are scaled up to multi-gram sample scale to evaluate scale-ability, solubility, and hygroscopicity. At this point, a finger print of pXRD, Near IR/Ramen or solid state NMR is obtained to positively identify the polymorphic form.
The determination of thermodynamic stable form follows via slurry experiment. Binary mixtures are slurried over days in solvents where the less stable and more soluble polymorph has the chance to convert to the more stable and less soluble polymorph. The sample is then scanned using pXRD to discern any polymorphic conversion. The more stable polymorph is then subject to additional slurry test using the animal vehicle to ensure no additional polymorphic changes such as conversion to the hydrated forms.
For the lead polymorph, the process is further scaled up and the crystallization conditions refined to bring about the desirable crystal habit. For instance, a change from fibrous to rod crystal habit is essential for the scale-up of the filtration process. At the same time, a solid state stability study preferably for the lead and the backup polymorphs, is initiated under accelerated storage conditions in order to confirm that the selected form is sufficiently stable to support a 2 year shelf-life.
Opportune Time to Select Salt/Polymorph
Although polymorph/salt screening should ideally be performed to select the optimum solid form upon selection of the lead compound prior to animal pharmacokinetic (PK) studies, these screening study can be costly and time consuming. But the consequences of late discovery of a thermodynamic form are grave, so there must be a strategy to minimize the risk without spending a large amount of resources. We find this right strategy based on early BCS classification of new compounds. We tailor the upfront polymorph/salt studies based on the risk in bioavailability, stability and manufacture-ability. Since regulatory agencies worldwide require the use of the same salt across preclinical and clinical studies, for insoluble or unstable compounds, salt screening is done early to enable further compound development. Once salt is selected, the polymorph screening of the selected salt if soluble may be done a little later after animal study. However it is paramount to confirm 1) the polymorph in use is stable in the toxicological vehicle, 2) no changes of solid forms during shipping and storage, 3) no significant degradation upon storage. Should there be polymorphic changes such as formation of a hydrate in the animal vehicle resulting in lowered solubility and precipitation of the hydrate, or formation of a hydrate when exposed to humidity during shipping and storage, early discovery of the stable forms will enable consistent animal exposure and avoid study repeats and delays in timelines. Therefore, although most companies do not perform comprehensive polymorph screening until late in the development cycle, we recommend identification of a thermodynamic stable form within the confine of not only the API manufacture processes but also in the designated animal and human formulations. For instance, for a drug product manufactured by direct compression, the solidstate properties of the active ingredient will likely be critical to the manufacture of the drug product, particularly when it constitutes the bulk of the tablet mass. On the other hand, for a drug product manufactured by wet granulation, the solidstate properties of the active ingredient may no longer be important but the potential for polymorphic conversion is high in the presence of high moisture contents. In the context of the effect of polymorphism on pharmaceutical processing, what is most relevant is the ability to consistently manufacture a drug product that conforms to applicable in-process controls and release specifications. This upfront work is especially critical to insoluble compounds prone to varied oral bioavailability in animal and human.
Early Pharmaceutical Development Cycle Time and API Utilization
Early pharmaceutical development cycle time is carefully synchronized between Discovery and Development to achieve a total of 12 months from lead compound declaration to IND filing. As shown in Figure 2, the first six months (Predevelopment Phase) are slated for evaluation of compound develop-ability. The second six months (Phase 0) are for INDenabling studies typically including one month toxicology studies in two animal species and clinical formulation development and clinical supplies manufacture. The evaluation of compound develop-ability encompasses pharmacology, pharmacokinetic, drug metabolism, toxicology, pharmaceutical and biopharmaceutical properties. For instance, a criteria for oral bioavailability is set at >20% for the first species and >10% for a second species.  For another instance, a criterion is set for stability profile of the compound to be sufficient to support a 2-year product shelf life. Prior to entry into the Predevelopment Phase, there is a PreSelection transition period for Discovery and Development to align the selections of solid form and animal formulation. In this interface from Discovery to Development, the choice of salt/polymorph is evaluated in conjunction of the targeted animal formulation to ensure seamless transition of pharmacology activities to pharmacokinetic profile. For example, Discovery may have been using an amorphous form giving higher animal exposure. The transition team is then charged to produce crystalline materials via salt formation or stable polymorph screening to bridge over the data from amorphous to crystalline materials. There are cases that the animal exposures is depressed below acceptable levels when changed from amorphous to crystalline form and the project is delayed pending Formulation department to provide a solubilized animal formulation to enable animal exposure for pilot toxicology studies.
For another instance, a criterion is set for stability profile of the compound to be sufficient to support a 2-year product shelf life. Prior to entry into the Predevelopment Phase, there is a PreSelection transition period for Discovery and Development to align the selections of solid form and animal formulation. In this interface from Discovery to Development, the choice of salt/polymorph is evaluated in conjunction of the targeted animal formulation to ensure seamless transition of pharmacology activities to pharmacokinetic profile. For example, Discovery may have been using an amorphous form giving higher animal exposure. The transition team is then charged to produce crystalline materials via salt formation or stable polymorph screening to bridge over the data from amorphous to crystalline materials. There are cases that the animal exposures is depressed below acceptable levels when changed from amorphous to crystalline form and the project is delayed pending Formulation department to provide a solubilized animal formulation to enable animal exposure for pilot toxicology studies.
The amount of API available to pharmaceutical development are 5 g for initial characterization and 25 g for form screening, both of which batches are prepared by discovery chemistry. The selected solid form is then scaled to 300g for the pilot toxicology studies. While the pilot study is on-going, a multi-kilogram GMP batch will be in preparation anticipating the compound successfully meeting the develop-ability criteria. The desire is to supply both the IND-enabling toxicology studies and the FIH clinical product with the same GMP API batch so that the impurity profile is consistent between the animal and human studies. However, it is not always feasible due to various reasons such as the insufficient quantity of starting materials or reagents. Sometimes, the time delay in further scale-up to a combined GLP/GMP batch is simply too much. Ideally, the 100g to 300 g for FIH formulation development should come from the GMP batch so that the performance can be directly scaled from laboratory to clinical batches. For a combined GLP/GMP batch, it is not a problem because the batch would be available upon IND lead declaration to start the GLP studies. However, for a scenario of split GLP and GMP batches, the GMP batch won’t be available until ~3 months into the phase 0 cycle. Careful comparison between the GLP and GMP batches would then be necessary to ensure success in scale-up of FIH formulation manufacture for clinical supplies.
BCS Based Decision Tree for Polymorph/Salt Selection
Since preliminary BCS classification is available through HTS pharmaceutical profiling, BCS can be used to rationalize the timing of polymorph/salt screening studies. A decision tree is presented in Figure 3 as an efficient way to prioritize these studies prior to animal work. This BCS-based decision tree is previously described in detail, together with a BCS-based animal formulation decision tree by the author [22]. For BCS 1 compounds, polymorphism or salt form is unlikely to impact on bioavailability and can be dealt with post animal studies. A salt form may still be of value if the physical properties or stability is improved for a BCS 1 compound. A typical scenario is to convert an oily free base to a free flowing powdered salt. On the other hand, for non-BCS 1 compound, the use of salt to increase solubility/bioavailability should be considered early. In Figure 3, “Soluble Salt” is defined as a salt form that enables the human dose soluble in 250 mL water. Should a soluble salt be discovered that provides improved bioavailability, animal PK studies may start prior to polymorph screening. Should the salt be still not soluble, the value of the salt would be questionable in consideration of the additional cost in the salt conversion step.
The dose volume of 250 mL is used herein in consistent with FDA BCS high solubility definition. However, for a highly permeable compound, a dose volume higher than 250 mL may still provide good absorption. It is especially true for compounds easily solublized by gastrointestinal surfactants. A dose volume as high as 5000 mL has previously been used as the breakpoint for selection of a solubilized dosage form for BCS class 2 compounds [6].  In the case that human dose can’t be estimated due to the novelty of the lead compound, the animal efficacious dose (ED) in mg/kg may instead be used with a dose volume of 100 mL/kg in the vehicle used in animal studies. Since compounds are usually given to animals in a volume of 10 mL/ kg, for a drug soluble in 100 mL/kg means 10% of the drug is in solution when given to animal. Solubility dependent dissolution is not likely to be the rate limiting step for absorption. The compound is considered soluble. For instance, compounds are considered soluble when ED = 10 mg/kg and solubility > 0.1 mg/mL, the dose volume = 10 / 0.1 =100 mL/kg or when ED = 1 mg/kg and solubility > 0.01 mg/mL. This oral potency dependent solubility gate criteria is implemented for entry into Predevelopment phase in author’s company
In the case that human dose can’t be estimated due to the novelty of the lead compound, the animal efficacious dose (ED) in mg/kg may instead be used with a dose volume of 100 mL/kg in the vehicle used in animal studies. Since compounds are usually given to animals in a volume of 10 mL/ kg, for a drug soluble in 100 mL/kg means 10% of the drug is in solution when given to animal. Solubility dependent dissolution is not likely to be the rate limiting step for absorption. The compound is considered soluble. For instance, compounds are considered soluble when ED = 10 mg/kg and solubility > 0.1 mg/mL, the dose volume = 10 / 0.1 =100 mL/kg or when ED = 1 mg/kg and solubility > 0.01 mg/mL. This oral potency dependent solubility gate criteria is implemented for entry into Predevelopment phase in author’s company
since 2004.
In conclusion, polymorph screening should be done prior to animal PK studies for non-BCS 1 compounds irrespective of free or salt form since different polymorphs are likely to impart different bioavailability. Conversely, animal studies may be performed prior to polymorph screening for BCS 1 compounds or a soluble salt of non-BCS 1 compounds since polymorphism is unlikely to impact on bioavailability. Adoption of a salt form should be based on the enhanced PK performance and/or manufacturability against the free form. We have used BCS-based decision tree described herein in the prioritization of salt and polymorph screening studies prior to animal studies for all NME since 2004. We have achieved selection of the right form >70% of the time prior to animal PK/TK studies and >90% of the time prior to IND-enabling toxicology studies. The right-first-time polymorph and formulation selection ensures consistent pharmacokinetic performance and avoids bridging BA/BE studies which is in line with FDA’s initiative to reduce R&D cycle time through quality by design for pharmaceutical product.
Conclusion
Looking into the future, we hope one day polymorphism of a chemical molecule may be comprehensively predicted through molecular modeling so that computer experimentation can replace the extensive laboratory works. In 2004, Price [23] and coworker reported a research initiative to develop computer software tools that consider the arrangement of atoms within a compound to predict whether that compound is likely to take on different crystal structures and if so, how many possible structures. This way a company may use such prediction to identify the most thermodynamically stable polymorph and tailor the manufacturing process in the production of the stable polymorph. Alternatively, the prediction may yield the conclusion that the polymorph at hand is stable within the confine of the storage environment and formulation presentations so to avoid upfront extensive screening for alternative polymorphs. Until such time, the BCS based polymorph/salt selection strategies can affect a minimum design and a higher efficiency with a lower cost.
In conclusion, it is not unusual that polymorphism issues have either delayed toxicological and clinical programs or disrupted commercial supply in the industry. Early selection of salt/polymorph enables a consistent pharmacokinetic exposure from one study to another right so that the development cycle may be compressed while securing the commercial intellectual property position. Salt/ polymorph screening has become an important activity in most pharmaceutical companies. The overarching goal is to select a thermodynamically stable, formulate-able and bioavailable solid form right the first time prior to any animal studies so that a 4 year cycle time from IND to NDA may be realized in line with FDA’s risk based initiative for the 21st century.
Acknowledgement
The author likes to thank Drs. Lisheng Kang and Marc Tesconi for their work on the Solid Steering Committee in Wyeth Research.
References
1. Amidon G. L., Lennernas H., Shah V. P., and Crison J. R. A theoretical basis for a biopharmaceutics drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 12(1995) 413–420.
2. “Guidance for Industry, Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral dosage Forms Based on a Biopharmaceutics Classification System”, Food and Drug Administration, Rockville, MD, 2000.
3. Wu Chi-Yuan and Benet, Leslie Z. Predicting Drug Disposition via Application of BCS: Transport/Absorption/ Elimination Interplay and Development of a Biopharmaceutics Drug Disposition Classification System. Pharm. Res. 22(2005)11–23.
4. Kerns E. H., Di L. Multivariate pharmaceutical profiling for drug discovery. Curr. Top. Med. Chem. 2(2002)87-98.
5. Byrn, S. et al “Pharmaceutical Solids: A Strategic Approach to Regulatory Considerations”, Pharm. Res., 12(1995) 945-954.
6. Chemburkar S.R., Bauer J., Deming K., Spiwek H., Patel K., Morris J., Henry R., Spanton S., Dziki W., Porter W., Quick J., Bauer P., Donaubauer J., Narayanan B.A., Soldani M., McFarland D., McFarland K., Dealing with the impact of ritonavir polymorphs on the late stages of bulk drug process development, Org. Process Res. Dev. 4(2000) 413–417.
7. Ruffolo, R. R., R&D Productivity: A New Way of Working At Wyeth, American Pharmaceutical Review, Volume 7, June 2004.
8. Lipinski C.A., Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev. 23 (1997) 3 – 25.
9. Lindenberg M., Kopp S., and Dressman J. B., Classification of orally administered drugs on the World Health Organization Model of Essential Medicines according to the Biopharmaceutics classification system, Eur. J. Pharm. Biopharm. 58(2004)265–278.
10. Ku M.S., An oral formulation decision tree based on the biopharmaceutical classification system for first in human clinical trials. Bulletin Technique Gattefosse 99 (2006):89-102.
11. Lipinski C.A., Poor aqueous solubility—an industry wide problem in drug discovery, Am. Pharm. Rev. 5 (3) (2002) 82– 85.
12. Venkatesh S., Lipper R.A., Role of the development scientist in compound lead selection and optimization, J. Pharm. Sci. 89 (2000) 145– 154.
13. Bighley L.D., Berge S.M., Monkhouse D.C., Salt forms of drugs and absorption, in: Swarbrick J., Boylan J.C. (Eds.), Encyclopedia of Pharmaceutical Technology 13,Marcel Dekker, New York, 1995, pp. 453– 499.
14. Yoshioka S., Stella V.J., Stability of Drugs and Dosage Forms, Kluwer Academic Publishers, Plenum Publishers, NY, 2000.
15. Guillory J.K., Poust R.I., Chemical kinetics and drug stability, in: Banker G.S., Rhodes C.T. (Eds.), 4th ed., Modern Pharmaceutics. Drugs and the Pharmaceutical Sciences 121, Marcel Dekker, New York, NY, 2002, pp. 139– 166 Revised and expanded.
16. “Guidance for Industry, ANDAs: Pharmaceutical Solid Polymorphism”, draft, Food and Drug Administration, Rockville, MD, 2007.
17. International Conference on Harmonization Q6A Guideline. Specifications for New Drug substances and Products: Chemical Substances, 1999 October.
18. Kwong, E.,”PAT Implementation Strategies for Pharmaceuticals –IQPC April 11-13, 2007.
19. Lang M.D., Grzesiak A.L., Matzger A.J., The use of polymer heteronuclei for crystalline polymorph selection, J. Am. Chem. Soc. 124 (2002) 14834–14835.
20. Morissette S.L., Almarsson Örn, Peterson M.L., Remenar J.F., Read M.J., Lemmo A.V., Ellis S., Cima M.J., Gardner C.R.. Highthroughput crystallization: polymorphs, salts, co-crystals and solvates of pharmaceutical solids. Advanced Drug Delivery Reviews 56 (2004) 275– 300
21. Almarsson, O., Gardner C.R., Novel approaches to issues of developability, January 2003, 21– 26. http://www. currentdrugdiscovery.com
22. Ku M.S., Use of the Biopharmaceutical Classification System in Early Drug Development. The AAPS Journal 10 (2008) 208-12.
23. Price, SL, The computational prediction of pharmarceutical crystal structures and polymorphism. Advanced Drug Delivery Reviews 56 (23): 301-319, 2004—International Centre for Diffraction Data.
Dr. M. Sherry Ku is responsible in Wyeth Research for pharmaceutical development activities from discovery support, lead selection, phase 0 and IND submission to clinical proof of concept. She worked on over 100 new clinical leads resulting in 78 initial IND filings. Sherry developed seven commercial products including Suprax, Zosyn/Tazocin, Zebeta, Isovorin, Thioplex, Sonata and Tygacil. She has a combined over 90 literature publications for patent and paper. She received a Ph. D. Degree in Pharmaceutics and Pharmaceutical Chemistry from The Ohio State University.