Abstract
During pharmaceutical development, the performance of pharmaceutical products intended for investigational purposes are often unknown and/or not optimized, however, one element critical to the safety and efficacy of investigational medicines is compliance with the cGMPs. Several mechanisms can be used to enhance and enable compliance in analytical laboratories where investigational medicines are tested and released for clinical use. The approach described here is an ‘in-house’ peer-led effort, which resulted in a program that has significant benefits and applicability for a dynamic, multi-disciplined analytical development department. The monitoring program aligns itself to both the Quality Systems approach [1] and the working practices of the laboratories; by tracking a randomly chosen sample from delivery, receipt, storage and analysis through to data reporting, notebook documentation, qualification and/or validation of instrumentation and electronic records. The program is based on peer-led laboratory monitorings (not audits) which help to distribute the responsibilities and benefits by increasing the overall awareness of the program. In this program, prominent issues can be quickly highlighted preventing propagation or escalation. The monitoring is information-rich and actionable metrics are gathered using a tracking form. This enables systematic problems to be highlighted and resolved on a department level versus repeatedly fixing the problem on a laboratory level. Critical involvement from both management and Quality Assurance (QA) ensures the credibility of the program.
Introduction
During pharmaceutical development, the performance of pharmaceutical products intended for investigational purposes are often unknown and/or not optimized, however, one element critical to the safety and efficacy of investigational medicines is compliance with the cGMPs. Several mechanisms such as notebook review, spot inspections, and varied training programs have been used to enhance and enable compliance in analytical laboratories where investigational medicines are tested and released for clinical use. To maintain and improve cGMP compliance in today’s analytical development environment, heightened awareness must be sustained. Programs such as annual GMP training and annual audits provide useful platforms for increasing awareness, but they do not provide ‘on the job’ training and therefore offer only a limited enhancement in maintaining quality compliance [2].
Earlier Manager Led Efforts
In the Analytical R&D laboratories at Bristol-Myers Squibb (BMS), an earlier compliance monitoring program used management representatives to perform regular ‘internal’ audits that focused on checking the level of compliance within individual laboratories. The program did not entail preparation of documents for review which in turn led to a cursory evaluation of laboratory compliance.
The original program combined general laboratory safety items, along with basic SOP adherence, cursory notebook review and confirmation of instrument calibration posted dates. The program lacked preliminary document retrieval, preparation and a point of focus, and unintentional deviations often went unnoticed. As shown in Figure A, the original program metrics gave a general overview of findings, but lacked the details necessary to drive improvements and lessons learned. For example, while it was clear that >40% of the findings were SOP related, it was not clear whether the problems were associated with one particular SOP or the currency of that SOP. This program did provide an advantage in that it gave management first-hand exposure to compliance issues in the laboratories, which allowed them to respond directly to problems identified. However, other areas of the program, such as the significant time commitment required by the management team to conduct the audits, the limited pool of auditors (which in turn reduced the frequency of audits), and the perception by bench scientists that the results of the audits may be punitive in nature, were disadvantageous.

Gap Analysis
From a review of the process, certain areas of opportunity for improvement were considered. The four main areas were:
- The program could benefit from utilizing the analytical chemists who have the hands-on knowledge and routine use of the laboratory tools to track, analyze and report sample results, including instrument and instrumental software packages and laboratory LIMS systems
- A thorough evaluation of the workflow and systems used in the laboratory is required when emulating the Quality Systems [1] approach
- Scheduling to coordinate the audit with laboratory personnel at a mutually convenient time is necessary to enable the maximum benefit from an “audit”
- Although time consuming, the collation and distillation of detailed information obtained from the “audit” is required to present a comprehensive picture of compliance within a department
A peer led new monitoring program that combined some aspects of the management only audit with more depth, being administered consistently, objective in nature and probed systems or practices to the depth required to assess quality operations, would be of significant value.
A Fresh Approach
As a solution, a strategy was devised that redistributed the time commitment across the entire department and provided a peer-led, one-on-one compliance training opportunity. This improved organizational efficiency and also led to other significant benefits that will be outlined later in this article. The design of the new program reduced management’s role to remove the “bigbrother” aspect of the original audit program and supplemented management with analytical scientists to lead the compliance monitoring. The benefits of peer monitoring avoid confrontational aspects of an audit and significantly increase the pool of participants from all levels of experienced personnel, and therefore increase the flexibility and impact of the program. The approach, which is a real time lessons-learned system, is more effective than a conventional training presentation. As peers evaluate the work of their colleagues, they indirectly re-evaluate their own practices and knowledge of procedural documents.
Program Foundations
The new program was designed to align itself to both the Quality Systems approach [1] and the working practices of the laboratories, with the goal of assessing compliance by means of tracking a previously random chosen sample, for release or stability testing, from receipt and storage, through analysis and data reporting. As electronic systems (such as LIMS, electronic databases and data acquisition software) are an integral piece of a scientist’s job function, an effective monitoring activity should include an assessment of the individual’s ability to utilize systems properly. The systems incorporated chromatography data management system, LIMS, template Model Documents, Document Repository and electronic training documents.
The approach entails checking notebook documentation, all instrumentation used in analysis (calibration records and cross references), laboratory operations, method execution and electronic records. The compliance of all key actions and documents is challenged and assessed against current expected departmental work practices, Instrument Operating Procedures (IOP’s), and Standard Operating Procedures (SOP’s) and is captured using a standard form. In order for the program to realize sustainable success, the program required significant planning and structural organization in several key areas:
- Alignment with both management and the responsible Quality Assurance unit
- Establishment of a Core Team to develop the program components, including:
o A universal tracking form to facilitate a focused 1 hour monitoring
o A monitoring guideline for lab monitors
o A comprehensive training program for lab monitors
o Recruitment of a monitoring team, consisting of membership from all levels of the department (lab technician to department director)
o A schedule for monitorings, including all relevant personnel and laboratories
o A mechanism for addressing issues / findings
o Review of findings from the monitoring along with associated metrics
o Presentation of findings / proposals to management

Program Overview
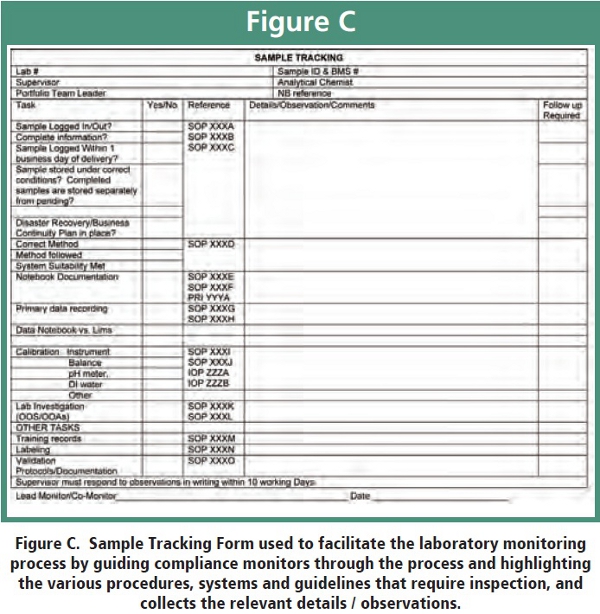
The release or stability sample selected for tracking during the monitoring is one that was analyzed by the selected personnel within the preceding 3-6 months. A schematic overview of the program schedule is outlined in Figure B with a chronological workflow of the lab systems monitoring outlined in Figure C. The samples selected are primarily clinical batch release or stability samples, although method validation [3] data may also be used if no stability or release batches are available. To enable the effective and efficient monitoring of these samples and ensure consistency, as the role of monitor rotates through the department, a sample tracking form (Figure D) was developed. It acts as a guide for the monitoring team to track a sample through the laboratory testing process, resulting in a focused, time-efficient review. The form also ensures that details are rigorously checked in a consistent, systematic and organized manner, and highlights the appropriate departmental SOPs, software packages, databases and departmental guidelines needed for review. The collated data is easily transferred to an electronic database, which, in turn, can be mined using standard searches to generate important departmental metrics. Collectively, this database represents the “hard evidence” of the department’s compliance performance including its deficiencies. The database provides not only a single laboratory view but also a view of the department as a whole when evaluating a specific period of time (e.g., one year).
The laboratory monitorings are performed using a lead and co-monitor, along with a relevant scientist from the laboratory being monitored. The lead and co-monitor select a random sample from the analyst’s laboratory sample logbook, schedule a mutually agreeable time for the monitoring with the laboratory scientist and retrieve pertinent documents such as the analytical methods used for analysis (e.g. HPLC assay, GC volatile impurity assay, dissolution assay, etc.), instrument operation procedures, analytical request forms (sample submission paperwork), specifications and LIMS data reports prior to the monitoring. Any findings observed during the monitoring are discussed with the laboratory scientist(s) prior to leaving the inspected laboratory. All findings are documented, and the completed tracking form is circulated to the scientist and supervisor by e-mail. Written response (if required) is expected within 10 days of receiving the documented findings and, upon resolution of any findings, the completed tracking form is archived.

Initially, pilot laboratory monitorings were performed by the ‘Core Team’ members, findings were evaluated and the program was then refined. The results and the final program were then presented for management endorsement.
The Quality Assurance (QA) unit was consulted regularly during the design of the peer-led program to ensure appropriate inspection components were incorporated into the program. Endorsement by management was critical to ensure participation by department members and commitment from the Monitoring Team. This established the triangle of critical relationships between the Core Team and the monitoring team, management and QA as illustrated in Figure E.
Program training was provided to the department and stressed that the new program was not designed to police the labs but enable monitors and lab personnel to partner, maximizing the potential impact. Initially, the Monitoring Team consisted solely of individuals who were provided one-on-one training by members of the Core Team during the course of a laboratory monitoring event. In turn, the co-monitors led subsequent monitorings expanding the pool of participants. 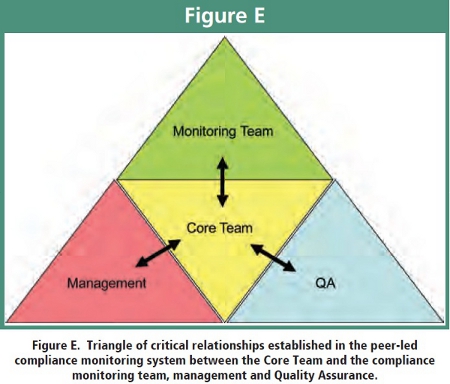 The long term objective of this approach is to systematically rotate the monitoring responsibilities throughout the department while providing on the job training with a goal to engage everyone in the department within a three to five year period. This will allow both the Monitoring Team and the scientists being monitored to complete a compliance evaluation of the laboratory and/or the overall department on a regular basis. This, in turn, reinforces the observance of cGMPs within the laboratories and increases overall compliance awareness.
The long term objective of this approach is to systematically rotate the monitoring responsibilities throughout the department while providing on the job training with a goal to engage everyone in the department within a three to five year period. This will allow both the Monitoring Team and the scientists being monitored to complete a compliance evaluation of the laboratory and/or the overall department on a regular basis. This, in turn, reinforces the observance of cGMPs within the laboratories and increases overall compliance awareness.
Added Benefits
As described earlier, peer-led compliance monitoring provides the benefit of wide-spread departmental involvement, significantly smaller demands on management time, and enables more monitoring events in a given period (a total of ~90 per year), which results in a comprehensive coverage of the department. In addition, the summarized monitoring information from the compliance database provides an objective assessment for management. This enables the differentiation between issues with working practices in the department, where a collective review of SOP / guidelines may be indicated, or issues with an isolated specific person or lab practice, where individualized training may be the most appropriate course of action. A key program benefit is that it provides a forum to discuss legitimate compliance concerns (an activity that is not a realistic option during an audit). In a management-led program, this is less likely to occur because of the perceived punitive nature of such an approach. In a peer-led monitoring program, the emphasis is shifted to one of verifying work practices in a less threatening atmosphere, prompting open, fruitful discussions that may not only highlight additional potential problems, but may also lead to potential solutions, as well.
Driving Changes
The Core Team members hold monthly meetings to discuss recent monitoring findings. This meeting provides a platform for experienced scientists to legitimately discuss compliance issues in the department, and propose solutions, in an in-depth manner. Over time, the team members are able to identify, not only the root of the compliance issue, but are able to utilize their insight from their daily laboratory responsibilities to suggest sensible and practical solutions to many of the findings uncovered during the monitorings. The collated compliance findings, metrics and the collective opinions of the Core Team members are used for quarterly presentations to management to highlight issues requiring resolution. This effort provides a more efficient engine to drive improvements in laboratory compliance. In turn, management has an objective view of compliance and can act on findings appropriately, and with confidence. A revised quarterly presentation, following management input and approval, can be used to provide feedback to the department at team meetings where further discussions on work practices and concerns are encouraged. Consequently, any changes to work practices are more likely to be understood and embraced by those in the laboratory. Alternatively, training programs in specific areas of deficiency may be suggested for the entire department, select groups or individuals. For the more significant issues identified, if they are deemed
a quality event, a corrective and preventative action (CAPA) may also result.
A continuous performance evaluation of the program is necessary to identify any progressive improvements needed to enhance the overall level of compliance in the department. These incremental changes are viewed as a positive evolution of the compliance program. This allows flexibility in the program to incorporate both changes driven by business needs or the regulatory landscape, enabling the program to stay fresh, innovative and in touch with the most current Quality Systems approaches. In general, monitoring versus auditing approach has had a direct impact on improving and maintaining compliant work practices in the department.
Over the two years since the program has been implemented, clear benefits have been realized:
- Laboratory personnel feel that the monitoring program is open, promotes discussion and is not an auditing or policing program to which they have little input.
- Laboratory personnel are more informed and have a more consistent understanding of compliance related expectations.
- Regular quarterly feedback to department with management endorsement has promoted awareness and reinforcement of the program.
- Continuous improvement to overall department compliance as illustrated by changes in work practices and refinement of SOPs.
- Validation of the program with close correlation with internal QA/GMP audit observations and internal comprehensive notebook reviews of all clinical batches released in 2004.
- Revision and improvements to the tracking form to reflect new observations and/or to further focus the monitoring.
Performance Discussion
Trending of the findings collected from the tracking forms for the first two years of the program showed (Figure F) a 30% drop in findings over the two-year interval. The most significant decreases were seen in the areas of notebook documentation errors and reagent labeling. However, from the detailed data (Figure G) it was clear that maintenance of instrument logbooks is an area where problems have continued to persist despite the implementation of the monitoring program. As a result, specific examples of notebook deficiencies have been given to QA and included in biannual trainings, IOP’s were reassigned for review, and regular logbook requirement discussions were added to monthly group meetings. Another highlighted area of concern was the inconsistent use of chart recorders to monitor cold rooms, refrigerators and freezers. Here a specific corrective and preventative action was taken by management to pursue the implementation of an automated computer based monitoring system that eliminated the paper based chart recorders, and, along with it, the majority of the compliance issues associated with the monitoring of the temperature. Overall, the data demonstrates a progressive improvement across the department although the variability from lab to lab “hazes” the picture. Analysis of the data is further complicated by an influx of new employees, and dynamic revisions of the monitoring program to address more stringent regulatory requirements, and to hone in on specific areas.
A concern that arose during the initial implementation of the pilot monitorings was the monitorings would take too much time to complete. However, it has become clear that the invested time, not only reduced the number of findings, but also led to time savings downstream and a greater confidence that the cGMP requirements were being followed in the laboratories. Based on its systematic approach, similar monitoring programs may be implemented in other areas of analytical development, such as, Registrational Stability testing, contract laboratories and process chemistry support.

Conclusions
A viable, succinct program that is flexible, routine, and sustainable and tailored to pharmaceutical development work practices has been described. The hallmarks of this program include membership from all levels of the department, a defined tracking form, an efficient one hour monitoring process, and the clear presentation of findings. The program captures the spirit of the Quality Systems based approach by tracking a sample from submission through data reporting. The program is an excellent solution for maintaining compliance in today’s versatile cGMP analytical laboratory environment, that serves both the laboratory personnel’s needs and the department’s as a whole.
Acknowledgements
We are grateful to Dan Carney, Tom Raglione, Joel Young, Anne Aubry, Leticia Quinones, Steve Klohr, Pankaj Shah, Barry Scheer, and the Compliance Monitoring Team for their contributions to this article.
References
1. US Food and Drug Administration, guidance for Industry: Quality Systems Approach to Pharmaceutical Current Good Manufacturing Practice Regulations, Draft guidance (FDA, Rockville, MD, 2004).
2. Code of Federal Regulations, Title 21, Food and Drugs, “Current Good Manufacturing Practice for Finished Pharmaceuticals” (General Services Administration, Washington D.C. Revised March 2005), Parts 210 and 211. Code of Federal Regulations, Title 21, Food and Drugs, “Quality System Regulation” (General Services Administration, Washington D.C. Revised March 2005), Part 820.20.
3. International Conference on Harmonization, “Validation of Analytical Procedures: Methodology,” Q2B, November 1996
Peter Tattersall is a Senior Research Investigator in Analytical Research and Development at Bristol-Myers Squibb Company in New Jersey. He received his BSc. and Ph.D. from the University of Manchester, UK. He worked in Analytical Development at AstraZeneca, Wilmington prior to joining Bristol- Myers Squibb in 2003 where he currently supports both API and drug product development.
Michelle Kubin is a Research Scientist in Analytical Research and Development at Bristol-Myers Squibb. She pioneered the peer-led compliance monitoring initiative within the department in 2002 and served in a leadership role for the team for five years, in addition to her bench level analytical responsibilities. She received her BSc. in biology and chemistry at Rider University. She worked in the Environmental Services Gas Chromatography Laboratory at Mobil Research, Pennington, prior to joining Bristol-Myers Squibb in 1997 where she currently supports both API and drug product from early development stages through NDA filing.
Anwar Hussain graduated with a BA in biochemistry from Rutgers Universtiy (1989), and obtained his MS in medicinal chemistry from the University of North Carolina at Chapel Hill (1996). He has worked in the pharmaceutical industry as a research scientist for over 10 years (over 9 years at Bristol-Myers Squibb in Analytical R&D). Currently he is pursuing a Masters in Business Administration from the University of North Carolina at Chapel Hill (Kenan Flagler Business School).