Introduction
Monoclonal antibodies (mAbs) can express their inherent physical instability by following one of several degradation pathways: adsorption, denaturation, or aggregation [1,2]. The most common physical degradation product for the majority of monoclonals is the formation of aggregates. Aggregates may have low solubility, reduced biological activity, and greater immunogenic potential. The presence of aggregates can make a product clinically unacceptable. During manufacturing, aggregates can lead to low recovery yields, and longer processing times. Antibody aggregation is thus one of the greatest challenges in developing antibody drugs. Efficient solutions to mAb aggregation require the understanding of not only aggregation mechanisms but also biophysical properties of aggregated material.
Aggregation can be influenced by factors such as concentration, viscosity, ionic strength, pH, and temperature. There are also various conditions that can lead to aggregation, among which the most frequent are mechanical stress [3] (pumping, filtration, mixing, or shaking), storage, freeze-thaw, and adsorption to surfaces/ interfaces. Depending on the solution conditions, degradation mechanisms can lead to native or denatured, soluble or insoluble aggregates. Insoluble aggregation of mAbs shaken in solution can produce both fibrils (at interface) and particles (in solution) [4]. The detailed mechanism of aggregate formation is unclear. Various techniques have been used to identify and quantify the soluble and insoluble aggregates; however, limited data is available on the characteristics of antibody aggregates [5,6,7].
Here we describe the results of a study that employs biophysical characterization in a kinetic approach to particle formation. The study was performed under conditions that promote particle formation in a mAb solution (a combination of buffer system and mechanical stress) where the protein is destabilized but not denatured. The biophysical characterization of the particles formed over the time course of the study was performed by using differential scanning calorimetry (DSC) [8,9,10] to evaluate the thermal stability of the mAb in solution, circular dichroism (CD) [5,6,8,11] to assess potential perturbation of secondary and tertiary structures associated with particle formation, intrinsic tryptophan (Trp) fluorescence to probe tertiary structure and, more specifically, the environment around aromatics, and finally, extrinsic 1-anilinonaphthalene-8-sulfonic acid (ANS) fluorescence [11,12] to asses solvent accessibility of hydrophobic clusters on the mAb surface. The biophysical data is further correlated with various analytical methods for particle and aggregates detection, and a particle formation mechanism is discussed.
Particle Formation in a mAb Solution
Product-related particles were identified in a citrate-salt solution formulation of a mAb during shear stress studies, long-term storage, and product vial filling. The filling process involved a recirculation step. In order to understand the nature and kinetics of particle formation, a recirculation study was performed that mimicked the vial filling process. The concentrated protein solution was placed in a reservoir and recirculated from the reservoir through the tubing using a pump set at 200 rpm. Additional shear stress was added by having a stir bar spin in the bottom of the reservoir. Aliquots were taken and testing performed at 0, 2, 6, 24, 30, 48, 54, and 72 h during recirculation.
Analytical Detection of mAb Particles
General appearance testing performed over the course of the study showed increased turbidity as a function of recirculation time. Solutions were initially clear, colorless, and free of visible particles; however, the 6 h samples contained visible particles, with the particle number significantly increasing after 24 h. The sub-visible particle count also increased drastically and rapidly (7-fold after 2 h, and 61–fold after 6 h, data not shown), reaching detector saturation after 24 h. In agreement with particle quantitation, solution turbidity (as measured by absorbance at 350 nm) also increased rapidly as a function of time. However, there was not a similar increase in soluble aggregate formation as monitored by size exclusion chromatography. Dynamic light scattering data indicated a mainly monomeric solution in the 0, 2, and 6 h samples; starting with the 24 h sample and above no data was acquired as the solution turbidity saturated the instrument. Despite the formation of a large number of visible and sub-visible particles, there was no significant change in protein concentration, indicating that the fraction of the protein forming particles was very small even when the solution became very turbid.
Thermal Stability of Particle Solutions by DSC
Knowledge of thermal stability of a protein drug in solution is typically achieved by measuring Tm, the “melting temperature”. The DSC thermograms of mAb solutions at various recirculation timepoints are presented in Figure 1. There is consistency between scans, with three unfolding transitions being clearly identified. From the four scans corresponding to the 0, 24, 48, and 72 h retains, average values of 68.8 ± 0.1ºC, 71.3 ± 0.0ºC, and 82.4 ± 0.1ºC were obtained for Tm1, Tm2, and Tm3, respectively. The average deviations among tested particle samples are no larger than the instrument precision, i.e. 0.2ºC. This is indicative of no changes in the tertiary fold, thermal stability of the protein, or thermodynamic populations.
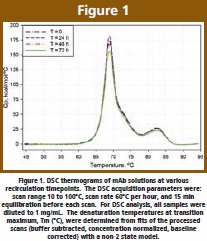 One interesting observation is the decrease in total area at 72 h. The decrease is not large (< 10%), however, it can be clearly identified. The reduction could suggest decreased availability of (active) folded protein, possibly due to lower availability of the monomer for thermal unfolding; at 72 h more protein is found in the associated (particle) state, thus less protein would be contributing to the total energy of unfolding.
One interesting observation is the decrease in total area at 72 h. The decrease is not large (< 10%), however, it can be clearly identified. The reduction could suggest decreased availability of (active) folded protein, possibly due to lower availability of the monomer for thermal unfolding; at 72 h more protein is found in the associated (particle) state, thus less protein would be contributing to the total energy of unfolding.
CD Evaluation of Structural Changes Associated with Particle Formation
The far-UV CD spectrum (Figure 2A) is that of a typical antibody, with a pronounced minimum at 217 nm indicative of high β-sheet content. There is also a positive shoulder at 235 nm, previously assigned to a combination of β-sheets and β-turns, and/ or to optically active (asymmetric) environments for aromatics [10]. During recirculation, the global secondary structure, as characterized by the 217 and 235 nm features, remains relatively unchanged.
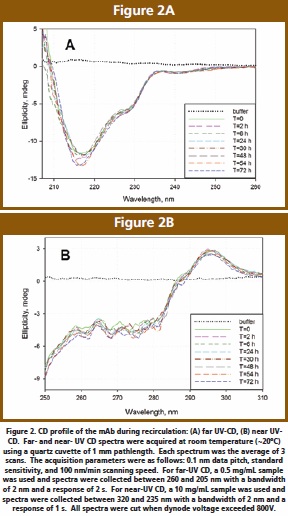 The near-UV CD spectrum (Figure 2B) is similar to the previously published scans [13] and includes complex contributions from side chains, aromatics and disulfide bonds. As previously described, the signal variation in 255 – 280 nm region can be assigned to minor asymmetries and/or altered mobilities in the aromatics environment. Overall, there are no significant changes in the immediate environment of active chromophores.
The near-UV CD spectrum (Figure 2B) is similar to the previously published scans [13] and includes complex contributions from side chains, aromatics and disulfide bonds. As previously described, the signal variation in 255 – 280 nm region can be assigned to minor asymmetries and/or altered mobilities in the aromatics environment. Overall, there are no significant changes in the immediate environment of active chromophores.
Detection of Particles by Intrinsic Trp Fluorescence
The fluorescence mainly due to Trp residues was selectively measured by excitation at 295 nm (Figure 3A). The wavelength to maximum fluorescence intensity (λmax) is 342 ± 1 nm, indicative of a partly-buried Trp, and pertaining to the spectral form II class (indole chromophore at the protein surface, in contact with bound water and other polar groups) [14]. The Trp peak position does not change during recirculation, suggesting that there is no structural perturbation or loss of globular packing associated with aggregate formation. This is in disagreement with previous studies [7] where significant red shifts in Trp fluorescence were observed, indicative of a partial reorganization of tertiary structure; in this published case, however, aggregates were formed during prolong exposures to siliconized glass.
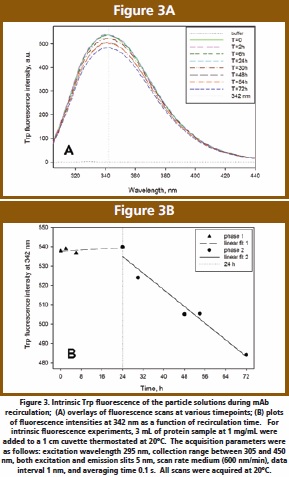 Intrinsic fluorescence intensity appears to be sensitive to particle formation, either through aggregate-induced light scattering contributions to the spectra, or through the aromatics-sensitive detection of local structural changes. During recirculation time there is a small (< 10%) decrease in the Trp signal (as measured by the fluorescence intensity), which indicates decreased hydrophobicity around this residue [6]. An increase in hydrophobicity is typically expected when the residues are located at the oligomer interface. This observation may suggest that the aromatics may be situated away from the interface between associated monomers and quenched by an increased exposure to solvent. However, not only exposure to aqueous environment but also almost all protein groups can quench the Trp signal to some extent [14]. Accordingly, the Trp may still be located at the multimer interface but internally quenched by one of these groups rearranged in the tertiary structure.
Intrinsic fluorescence intensity appears to be sensitive to particle formation, either through aggregate-induced light scattering contributions to the spectra, or through the aromatics-sensitive detection of local structural changes. During recirculation time there is a small (< 10%) decrease in the Trp signal (as measured by the fluorescence intensity), which indicates decreased hydrophobicity around this residue [6]. An increase in hydrophobicity is typically expected when the residues are located at the oligomer interface. This observation may suggest that the aromatics may be situated away from the interface between associated monomers and quenched by an increased exposure to solvent. However, not only exposure to aqueous environment but also almost all protein groups can quench the Trp signal to some extent [14]. Accordingly, the Trp may still be located at the multimer interface but internally quenched by one of these groups rearranged in the tertiary structure.
Although the quenching of Trp signal over time is small, it would be instructive to establish if a trend can be detected. Thus, when the fluorescence intensity at 342 nm is monitored as a function of time (Figure 3B), a biphasic quenching is observed. The emission signal remains constant for the first 6 to 24 h, suggestive of a lag time and correlating with a clear protein solution. Subsequently, past the 24 h timepoint, the Trp signal decreases and aggregates are now visible (as described in the analytical detection section). This second phase could correspond to oligomer formation to a point where solubility limit is reached and the associated protein is visible in solution as particles.
Detection of Particles by Extrinsic ANS Fluorescence
ANS is a dye whose fluorescence is greatly enhanced on binding to hydrophobic surfaces. ANS transfer from a polar to a non-polar medium results not only in a blue shift (lower wavelength) in its maximum emission wavelength λmax, but also in a significant increase in its quantum yield [15].
In the absence of protein, ANS displays one emission maximum at 528 nm [16]. The ANS fluorescence intensities increase as a function of recirculation time (Figure 4A). Simultaneously, there is a drastic blue shift in ANS maxima with time. Blue shifts of 10 nm (initial time point) to 29 nm (last time point) in ANS fluorescence are observed upon binding to protein. This is in agreement with previous results [7,16], where shifts of 14 nm were observed for particulates.
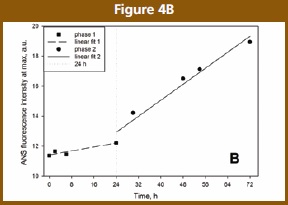
The emission wavelength was monitored. When the fluorescence intensity at λmax is plotted as a function of time (Figure 4B), the ANS detected self-association appears bi-phasic. Similar to Trp fluorescence, there are no significant changes in ANS fluorescence up to 24 h. This could correspond to a lag phase (slow kinetics, rate-limiting step), and may suggest a nucleation mechanism. Although ANS binding to the native state was weak, beyond 24 h the extent of ANS binding increased drastically in a size- and number- dependent manner in aggregates, as expected [5,17]. This could be consistent to a growth phase (to insoluble aggregation). In theory, a plateau is expected when the solution is depleted of monomer, creating a sigmoidal kinetics typical of protein aggregation and fibrillation; our timeframe does not cover this final stage of aggregation.
 Quality of mAb Solution After Particle Filtration
Quality of mAb Solution After Particle Filtration
At selected time points, the protein was filtered (through a syringe filter) and testing was performed as described above. It was found that the filtered solutions at various post-6 h timepoints had all analytical and biophysical properties similar to those of the protein at the start of the study (data not shown). This suggests that only a small fraction of the total protein is involved in particle formation, and that mAb association leaves the remaining protein in an unaltered native form.
Mechanistic Speculations
In this study, solution recirculation led to particles formation. Protein aggregation usually leads to loss of tertiary structure; however, in our study no changes were detected within the native state of the mAb. Clearly if there are subtle conformational aspects at work they escaped our biophysical detection [18]. Our data suggest mAb association of the native state or through a native-like subpopulation prone to aggregation. This idea is not novel [7,19,20]; however, the mechanistic details of self-association in native-like conditions remain understudied and poorly understood.
As detected by fluorescence, self-association appears biphasic and proceeds without any detectable structural changes in the pre-aggregated protein. Our interpretation of the analytical and biophysical data suggests that association occurs before major (detectable) structural changes are observed. The nature and the extent of self-association remain to be better defined. We hypothesize that initially the protein forms aggregation nuclei (detectable as sub-visible particles) during a lag phase. A nucleation mechanism was previously proposed for IgG2 [21]; similar to our case, no major structural changes occurred upon precipitation. During the lag time our mAb solution remains clear. The nuclei aggregate in the native state may then reach a certain size or undergo an undetectable conformational change that triggers the subsequent oligomer growth phase. Aggregates are now visible and there may be evidence of structural reorganization within the native-like intermediates [11]. The particles, however, constitute a small percent of the total protein, and their formation does not affect the quality of the protein remaining in solution post-filtration.
Summary
Stress and stability studies conducted on an antibody solution revealed this monoclonal’s propensity to form particles. Experiments were conducted to understand the nature and the mechanism of particle formation upon application of shear. Extensive analytical and biophysical characterization of particles suggests a native-state nucleation mechanism. Despite the formation of a large number of visible and sub-visible particles, the fraction of the protein forming particles was very small. The formation of particles does not impact the quality and quantity of the monomeric protein remaining in solution.
References
- Wang, W. Protein aggregation and its inhibition in biopharmaceutics. Int. J. Pharm. 2005, 289, 1-30.
- Wang, W.; Singh, S.; Zeng, D. L.; King, K.; Nema, S. Antibody structure, instability, and formulation. J. Pharm. Sci. 2007, 96, 1-26.
- Mahler, H. C.; Muller, R.; Friess, W.; Delille, A.; Matheus, S. Induction and analysis of aggregates in a liquid IgG1-antibody formulation. Eur. J. Pharm. Biopharm. 2005, 59, 407-417.
- Henson, A. F.; Mitchell, J. R.; Musselwhite, P. R. The surface coagulation of proteins during shaking. J. Colloid Interface Sci. 1970, 32, 162-165.
- McCarthy, D. A.; Drake, A. F. Spectroscopic studies on IgG aggregate formation. Mol. Immunol. 1989, 26, 875-881.
- Demeule, B.; Lawrence, M. J.; Drake, A. F.; Gurny, R.; Arvinte, T. Characterization of protein aggregation: the case of a therapeutic immunoglobulin. Biochim. Biophys. Acta 2007, 1774, 146-153.
- Souillac, P. O. Biophysical characterization of insoluble aggregates of a multi-domain protein: an insight into the role of the various domains. J. Pharm. Sci. 2005, 94, 2069-2083.
- Vermeer, A. W.; Norde, W. The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein. Biophys. J. 2000, 78, 394-404.
- Ahrer, K.; Buchacher, A.; Iberer, G.; Jungbauer, A. Thermodynamic stability and formation of aggregates of human immunoglobulin G characterised by differential scanning calorimetry and dynamic light scattering. J. Biochem. Biophys. Methods 2006, 66, 73-86.
- Harn, N.; Allan, C.; Oliver, C.; Middaugh, C. R. Highly concentrated monoclonal antibody solutions: direct analysis of physical structure and thermal stability. J. Pharm. Sci. 2007, 96, 532-546.
- Plakoutsi, G.; Bemporad, F.; Calamai, M.; Taddei, N.; Dobson, C. M.; Chiti, F. Evidence for a mechanism of amyloid formation involving molecular reorganisation within native-like precursor aggregates. J. Mol. Biol. 2005, 351, 910-922.
- Murali, J.; Jayakumar, R. Spectroscopic studies on native and protofibrillar insulin. J. Struct. Biol. 2005, 150, 180-189.
- Dani, B.; Platz, R.; Tzannis, S. T. High concentration formulation feasibility of human immunoglubulin G for subcutaneous administration. J. Pharm. Sci. 2007, 96, 1504-1517.
- Ladokhin, A. S. Fluorescence spectroscopy in peptide and protein analysis. In Encyclopedia of Analytical Chemistry; Meyers, R. A., Ed.; John Wiley & Sons Ltd, Chichester: 2000; pp. 5762-5779.
- Easterbrook-Smith, S. B.; Dwek, R. A. The use of ANS fluorescence as a probe for immunoglobulin flexibility. FEBS Lett. 1980, 121, 253-256.
- Kamyshny, A.; Magdassi, S.; Relkin, P. Chemically Modified Human Immunoglobulin G: Hydrophobicity and Surface Activity at Air/Solution Interface. J. Colloid Interface Sci. 1999, 212, 74-80.
- Lindgren, M.; Sorgjerd, K.; Hammarstrom, P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys. J. 2005, 88, 4200-4212.
- Chen, Y.; Ding, F.; Nie, H.; Serohijos, A. W.; Sharma, S.; Wilcox, K. C.; Yin, S.; Dokholyan, N. V. Protein folding: Then and now. Arch. Biochem. Biophys. 2007.
- Kendrick, B. S.; Carpenter, J. F.; Cleland, J. L.; Randolph, T. W. A transient expansion of the native state precedes aggregation of recombinant human interferon-gamma. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 14142-14146.
- Plakoutsi, G.; Taddei, N.; Stefani, M.; Chiti, F. Aggregation of the Acylphosphatase from Sulfolobus solfataricus: the folded and partially unfolded states can both be precursors for amyloid formation. J. Biol. Chem. 2004, 279, 14111-14119.
- Scoville, C. D.; Turner, D. H.; Lippert, J. L.; Abraham, G. N. Study of the kinetic and structural properties of a monoclonal immunoglobulin G cryoglobulin. J. Biol. Chem. 1980, 255, 5847-5852.
Sorina Morar-Mitrica received a B.S. in Chemistry and M.S. in Biocatalysis from Babes-Bolyai University in Romania, and her Ph.D. in Chemistry from the University of North Carolina at Chapel Hill. After graduating, she joined Diosynth Biotechnology where she worked on various aspects of protein drug development including purification, PEGylation, and preformulation. Sorina joined GlaxoSmithKline (GSK) in 2007 as an investigator within the Biopharmaceutical Technologies group, where she continues to work on formulation development of protein drugs – with a main focus on the biophysical characterization and high-throughput formulation.
Charlene Brisbane received her BS in Biological Sciences from Canegie Meilon University and MBA in International Business from Temple University. She has spent over 20 years in the pharmaceutical industry starting her career with Johnson & Johnson (J&J) where she had the opportunity to work on the purification, analysis, formulation and delivery of several biopharmaceutical compounds. Charlene joined GSK in 1996 and currently is an investigator in Biopharmaceutical Technologies where she continues to work on the formulation development of biopharmaceutical, as well as, technology transfer, device and delivery activities.
Douglas Nesta received his BA in Biology from Rutgers University and his MS in Biology from New York University. Doug joined the Medical research Division of American Cyanamid Company in 1984, where he was one of the initial members of the biopharmaceutical development department. During this period, he also completed his Ph.D. in Environmental Health Sciences at NYU. In 1995, Doug joined GSK Beecham R&D, as senior investigator within Pharmaceutical Technologies, first as a member of a biopharm product development team and then progressing to leadership roles in this department. For the last five years, he has served as director of the Biopharmaceutical Technologies department within GSK R&D, with overall responsibility for biopharm drug product formulation and dosage form development, as well as, development and optimization of secondary manufacturing processes and delivery technologies.
Amol Ketkar is a Manager in the Biopharmaceutical Product Development group in GSK. He obtained his B. Pharm degree from the University of Bombay, in India and his Ph.D. in Pharmaceutics from Ohio State University. Amol’s experience is primarily in the areas of parenteral formulation development and manufacturing. He currently leads a multi-disciplinary team focused on formulation development and drug delivery of biologics.