Abstract
Modern pharmaceutical development continues to move away from the trial-and-error method favored by Thomas Edison, and is moving toward a more computationally assisted approach that serves to minimize unnecessary experimental work. Since the formation constant of a salt species can be approximated knowing only the pKA value of the acid and the pKB value of the base, it is then possible to calculate the range of ionization constants of salt-forming agents that would ensure the formation of salts in high degrees of efficiency. Once the appropriate range of salt forms has been identified, one may study only those products predicted to have suitable properties. The utility of the methodology was demonstrated by predicting the existence of a number of robust salts of famotidine, and then through preparation and study of the famotidine salicylate salt.
Introduction
It is an unfortunate fact of life that active pharmaceutical ingredients frequently do not exhibit the range of physical properties that makes them directly suitable for development. One of the approaches that is used to modify the characteristics of drug substances is to employ a salt form of the substance, since salts enable one to modify aqueous solubility, dissolution rate, solution pH, crystal form, hygroscopicity, chemical stability, melting point, and even mechanical properties. The beneficial aspects of using of salt forms as active pharmaceutical ingredients is well known, and represents one of the means to increase the degree of solubility of otherwise intractable substances [1,2,3], and hopefully to increase bioavailability [4].
Traditionally, pharmaceutical scientists have favored the trialand- error method of Thomas Edison where one obtains a stock of drug substance and proceeds to make as many salt forms as there are salt-formers on the shelf. Once this work was conducted in lots of little beakers, and lately workers favor the use of 96-well plates [5], but the principle is the same. At the end of the screening process, one uses analytical methods to determine the products exhibiting acceptable degrees of aqueous solubility and dissolution rate, appropriate crystal form of low hygroscopicity, high melting point, good mechanical properties, and acceptable chemical stability to become the chosen candidates for further development. The only restriction usually incorporated in the design of these studies is that the salt-forming counterion must be one of the pharmaceutically acceptable species identified in compilations [1-3, 6].
There have been numerous attempts to use intelligent design during the salt selection process. Gould [7] used a decision analysis process to develop a rational process of salt selection for basic drugs, while Morris et al [8] described a more general method that was used to guide the salt selection of a drug candidate. The theoretical basis and application of in situ salt screening has been applied monobasic and monoacidic substances [9] and extended to cover multibasic drugs in multiprotic acids [10]. Procedures for salt selection and optimization have been reviewed [11], as have strategies for salt selection [12] and optimization of salt forms [13]. The use of a gridbased molecular modeling method for salt screening has also been described [14].
In a recent note, a consideration of the chemical equilibria associated with weak acids, bases, and their salts was used to derive a useful relation that enables one to approximate the formation constant of a salt form [15]. In the process, one first calculated an ionization constant for the drug substance under study, and then used the equation was used to calculate the degree of stability (or dispropionation) for salt-formers as a function of their ionization constant value. This approach facilitated the selection of what salt forms would be appropriate for development, which could then be isolated as studied further.
The Computational Aspect
The equation approximating the formation constant of a salt form has been derived previously [15], so only the briefest details will be provided here. In the Brønsted-Lowry model, an acid is a substance capable of donating a proton to another substance (such as water):
HA + H2O  H3O+ + A– (1)
H3O+ + A– (1)
and this reaction is characterized by the concentration-based equilibrium constant expression:
KA = [H3O+] [A–] / [HA] (2)
In the same model, a base is a substance capable of accepting a proton donated by another substance (such as water):
B + H2O BH+ + OH– (3)
whose concentration-based ionization constant expression is written as:
KB =[BH+] [OH–] / [B] (4)
Now, a salt is formed by the transfer of a proton from an acid (HA) to a base (B) capable of accepting that proton:
HA + B HB+ + A– + H2O (5)
A very interesting relation can be obtained by multiplying equation (2) by equation (4):
KA KB =[H3O+] [A–] [HB+] [OH–] / [HA] [B] (6)
But since the product [H3O+] [OH–] is the ionization product of water, one can rearrange equation (6) to yield:
KA KB / KW = [A–] [HB+] / [HA] [B] (7)
Equation (7) may be recognized as the equilibrium constant expression corresponding to the neutralization reaction of equation (5).
One can re-define the constants on the left-hand side of equation (7) to be the salt formation constant:
KS = KA KB / KW (8)
When converted to the Sørensen scale, equation (8) becomes:
pKS = pKA + pKB – pKW (9)
The ability to calculate KS (or pKS) enables one to estimate the relative position of the equilibrium described by equation (5). For example, if log(KS) = 2, it is easy to calculate that the neutralization reaction would proceed 90.91% to completion. Similarly, if log(KS) = 3, the efficiency of salt formation would be 96.93%, and if log(KS) = 4, the salt would be 99.01% formed.
The Empirical Aspect
The ability to calculate log(KS) values can be of great value in designing the scope of a salt selection study in that it can be used to determine the acidity or basicity range for potential salt forming species. To illustrate this principle, we will consider the case of famotidine, or 3-[[[2-[(aminoiminomethyl)amino]-4-thiazolyl] methyl]thio]-N-(aminosulfonyl)propanimidamide:
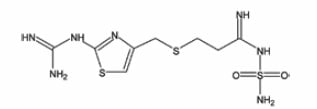
Some variability in ionization constants has been reported for this compound [16], but an average pKA value of 6.7 will serve for the present discussion. For this drug substance, pKB would therefore equal 7.3, and so the log(KS) values and salt formation efficiencies collected in Table 1 would be calculated for a series of acids having varying pKA values.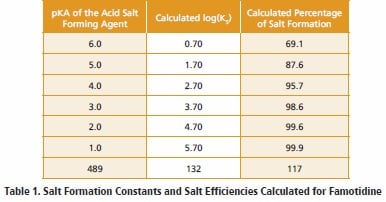
Using an acceptability threshold of 98-99% salt formation efficiency as indicating the existence of a salt form that would be characterized by an acceptably low degree of disproportionation when dissolved in water, one would conclude that only acids having pKA values less than 3.0 would form acceptable salt forms. With this conclusion in hand, one would then consult the compilations of pharmaceutically acceptable acids [1-3, 6] to identify possible candidates for that would then actually be prepared in the laboratory. A list of pharmaceutically acid candidates that would be predicted to form robust salts with famotidine is listed in Table 2, along with the computed parameters for the various salt forms.
The preparation of these salts could be effected by the simple dissolution of equimolar amounts of famotidine and the pharmaceutically acceptable acids of Table 2 in a suitable solvent (methanol, for example), and then crystallizing the product. These salts would then be evaluated on the basis of the degree of acceptability associated with their solubility, hygroscopicity, or other physical properties. For example, the tiered acceptability criteria outlined in the paper by Morris et al [8] could be used to design the program of salt form evaluation.
Case Study: Famotidine Salicylate
 As discussed in the previous section, since salicylic acid is known to have a pKA equal to 2.97, the log KS of the famotidine salt is calculated to be 3.73, and therefore one would expect that a salt that would form with a 98.65% efficiency would be acceptable for development. However, even though one would be absolutely certain from the calculations that the salt would form, the physical properties of the actually isolated salt form are not predictable.
As discussed in the previous section, since salicylic acid is known to have a pKA equal to 2.97, the log KS of the famotidine salt is calculated to be 3.73, and therefore one would expect that a salt that would form with a 98.65% efficiency would be acceptable for development. However, even though one would be absolutely certain from the calculations that the salt would form, the physical properties of the actually isolated salt form are not predictable.
To demonstrate this fact, equimolar amounts of famotidine and salicylic acid were dissolved in methanol, and the solution allowed to undergo slow evaporation. At the conclusion of this process, only a non-crystalline, viscous oil was obtained. However, this oil could then be triturated with methylene chloride, and a crystalline solid was obtained at the end of the process. This particular isolation procedure illustrates the value of making proposed salts on a larger scale, since one would not have obtained a crystalline solid through simple evaporation in a 96-well plate and that could have caused one to miss the opportunity of studying a promising salt form.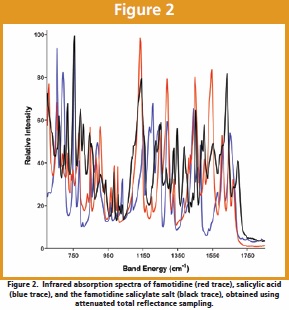
The famotidine salicylate salt was studied using a standard set of physical characterization methods. As shown in Figure 1, the X-ray powder diffraction pattern of the salt was found to be different and distinct relative to the initial reactants, demonstrating the existence of a new crystal structure. As shown in the Fourier transform infrared absorption spectra of Figure 2, the vibrational band at 1652 cm–1 associated with the carboxylic acid group of salicylic acid was not observed in the spectrum of the salt, having been replaced by the band at 1634 cm–1 associated with the analogous vibrational mode of the carboxylate anion.
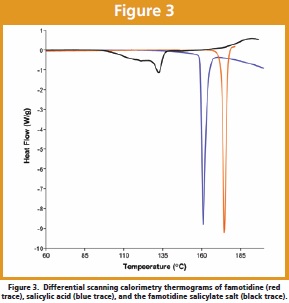 The differential scanning calorimetry thermograms of the two reactants and the salt complete the characterization. As shown in Figure 3, famotidine exhibits a melting endotherm having a maximum at 174.5ºC, salicylic acid a melting endotherm having its maximum at 160.9ºC, while the famotidine salicylate salt exhibited a complicated sequence of melting transitions, the most well-defined of which exhibited a melting endotherm having a maximum at 132.6ºC.
The differential scanning calorimetry thermograms of the two reactants and the salt complete the characterization. As shown in Figure 3, famotidine exhibits a melting endotherm having a maximum at 174.5ºC, salicylic acid a melting endotherm having its maximum at 160.9ºC, while the famotidine salicylate salt exhibited a complicated sequence of melting transitions, the most well-defined of which exhibited a melting endotherm having a maximum at 132.6ºC.
The obviously lower enthalpy of fusion for the famotidine salicylate salt relative to the enthalpies of fusion for the initial reactants coupled with its lower melting temperature suggests that the lattice energy of the salt form is considerably smaller than that of the reactants. This in turn would suggest that the aqueous solubility of the famotidine salicylate salt should be much larger than that of famotidine itself, which is approximately 0.1 mg/mL [17]. To verify this possibility, approximately 900 mg of the famotidine salicylate salt was found to dissolve in 40 mL of water, indicating an aqueous solubility exceeding 20 mg/mL.
Conclusions
The identification of a robust salt form of an active pharmaceutical ingredient is one approach to consider should the characteristics of the free acid or free base not be acceptable, and selection of an appropriate salt form of a drug substance provides one with the possibility to selectively modify a significant number of the physical properties of a drug substance. It has been demonstrated that one does not have to conduct salt screening studies following trial-and-error procedures, since appropriate saltforming candidates can be readily identified through knowledge of the ionic equilibria of the acids and bases involved. Beginning with the ionization constant (either calculated or measured) of the compound in question, one may use the salt formation constant expression to deduce the range of ionization constants of saltforming agents that would ensure the formation of salts in high degrees of efficiency and which would be stable with respect to disproportionation. Once a set of salt-forming agents is identified, the salts themselves would then be prepared in solid form for further characterization.
References
1. SM Berge, LD Bighley, and DC Monkhouse. Pharmaceutical Salts. J. Pharm. Sci., 66: 1-19 (1977).
2. BD Anderson and KP Flora. Preparation of Water-Soluble Compounds Through Salt Formation. Chapter 34 in The Practice of Medicinal Chemistry, CG Wermuth, ed. Academic Press, New York, 1996, pp. 739-754.
3. PH Stahl and CG Wermuth. Handbook of Pharmaceutical Salts. Wiley-VCH, Weinheim, Germany, 2002.
4. LD Bighley, SM Berge, and DC Monkhouse. Salt Forms of Drugs and Absorption. in Encyclopedia of Pharmaceutical Technology, J Swarbrick and JC Boylan, eds. Marcel Dekker, New York, 1995, pp. 453-499.
5. EC Ware and DR Lu. An Automated Approach to Salt Selection for New Unique Trazodone Salts. Pharm. Res., 21: 177-184 (2004).
6. DA Haynes, W Jones, and WDS Motherwell. Occurrence of Pharmaceutically Acceptable Anions and Cations in the Cambridge Structural Database. J. Pharm. Sci., 94: 2111-2120 (2005).
7. PL Gould. Salt Selection for Basic Drugs. Int. J. Pharm., 33: 201-217 (1986).
8. KR Morris, MG Fakes, AB Thakur, AW Newman, AK Singh, JJ Venit, CJ Spagnuolo, and ATM Serajuddin. An Integrated Approach to the Selection of Optimal Salt Form for a New Drug Candidate. Int. J. Pharm., 105: 209-217 (1994).
9. W-Q Tong and G Whitesell. In Situ Salt Screening – A Useful Technique for Discovery Support and Preformulation Studies. Pharm. Dev. Tech., 3: 215-223 (1998).
10. M Sacchetti. General Equations for In Situ Salt Screening of Multibasic Drugs in Multiprotic Acids. Pharm. Dev. Tech., 5: 579-582 (2000).
11. RJ Bastin, MJ Bowker, and BJ Slater. Salt Selection and Optimization Procedures for Pharmaceutical New Chemical Entities. Org. Proc. Res. Dev., 4: 427-435 (2000).
12. ATM Serajuddin and M Pudipeddi. Salt-Selection Strategies, chapter 6 in Handbook of Pharmaceutical Salts, PH Stahl and CG Wermuth, eds. Wiley-VCH, Weinheim, Germany, 2002, pp. 135-160.
13. MJ Bowker. A Procedure for Salt Selection and Optimization. chapter 7 in Handbook of Pharmaceutical Salts, PH Stahl and CG Wermuth, eds. Wiley-VCH, Weinheim, Germany, 2002, pp. 161-189.
14. RB Hammond, RS Hashim, C Ma, and KJ Roberts. Grid-Based Molecular Modeling for Pharmaceutical Salt Screening: Case Example of 3,4,5,7,8,9-hexahydro-2H-pyrimido-(1,2-a)- primidinium acetate. J. Pharm. Sci., 95: 2361-2372 (2006).
15. HG Brittain. Strategy for the Prediction and Selection of Drug Substance Salt Forms. Pharm. Tech., 31(10): 78-88 (2007).
16. RJ Prankerd. Critical Compilation of pKA Values for Pharmaceutical Substances. in Profiles of Drug Substances Excipients, and Related Methodology, HG Brittain, ed. Elsevier-Academic Press, Amsterdam, 2007, p 191.
17. MJ O’Neil, PE Heckelman, CB Koch, and KJ.Roman Famotidine; entry 3930. in The Merck Index. Merck & Co., Whitehouse Station, 2006, p 3935.
Dr. Brittain is presently the Institute Director of the Center for Pharmaceutical Physics, a research and consulting institute he established for the study of substances having pharmaceutical interest. Dr. Brittain received his Ph.D. in physical chemistry from the City University of New York, and has held full-time faculty positions at Ferrum College and Seton Hall University. Dr. Brittain has authored over 300 research publications and book chapters, and has presented over 150 invited lectures and short courses in the pharmaceutical field. He has edited the monographs Physical Characterization of Pharmaceutical Solids, Polymorphism in Pharmaceutical Solids (two editions), and Spectroscopy of Pharmaceutical Solids, and has also co-edited the monograph Preformulation in Solid Dosage Form Development. Dr. Brittain is an Associate Editor for Physical Pharmacy for the Journal of Pharmaceutical Sciences, and a member of the editorial boards of Pharmaceutical Development and Technology, AAPS PharmSciTech, and Pharmaceutical Technology. He is also the Editor for the book series Profiles of Drug Substances, Excipients, and Related Methodology.