Abstract
Karl Fischer titration (KFT) is the most widely used analytical technique for determination of water content in the pharmaceutical industry. Accurate determination of water content is not only important in understanding the performance of drug substances and drug products, but also a limiting factor to the accuracy of drug content calculated on anhydrous and solvent free basis. The variability of the KFT results, therefore, directly impacts on water and drug content specifications. In recognition of the importance of KFT method and the lack of availability of a science- and risk-based method development approach for KFT in a QbD regulatory environment, we introduce a comprehensive method development strategy (MDS) in this article. The discussion includes many examples to illustrate how QbD principles can be applied together with flowcharts to the development of robust KFT methodology.
Introduction
Water content is typically a registered specification test for both drug substances and drug products [1]. Karl Fischer titration (KFT), a selective and quantitative method for water determination, is the preferred technique in the pharmaceutical industry [2-5]. Water plays a critical role in physical and chemical stability of active pharmaceutical ingredient (API) and drug product (DP) [6]. Water can induce degradation of drug compound, cause undesired crystalline phase transitions, as well as lower glass transition temperatures for amorphous lyophilized products [7,8]. Additionally, water promotes microbiological activity which can have a major impact on the safety of the drug product [9-11].
KFT analysis is based on the modified Bunsen reaction, in which water reacts stoichiometrically with iodine in a suitable solvent and in the presence of an organic base [12]. Depending on how iodine is provided, KFT methods can be divided into two classes, namely volumetric and coulometric Karl Fischer titrations (vFKT and cKFT). In vKFT, iodine is formulated in the titration reagent; as opposed to being electrolytically generated in situ at the anode in cKFT. Samples can be introduced to the titration directly as solids, liquids, or pre-dissolved solutions. Alternatively, an oven accessory can be used, wherein water is thermally released and the resulting water vapor is transferred by inert gas stream to the titration cell. An oven accessory is typically integrated to a cKFT titrator, which accommodates compounds not amendable to direct titration [13] to overcome issues such as undesired side reactions and poor solubility of samples that may obstruct the electrode. As a result, oven KFT has evolved into an important method, especially for difficult sample matrices including in-process chemical samples [14], dairy products [15,16], and etc. [17-20]. Advances in KF instrumentation combined with improved quality of commercial titration reagents [21,22] have led to reliable routine analysis. The maturity of KFT is evident by recognition of various pharmacopoeia or standard testing methods, including vKFT in USP<921> Method Ia/Ib, and ASTM E203; and cKFT in USP<921> Ic and ASTM E1064. Nevertheless, the provisions and caveats are less scrutinized during method development and validation, e.g., cKFT assumes 100% coulometric consumption by water only. Additionally, the nature of the sample may prevent interchangeable use of vKFT or cKFT. Working principles for common KFT methodologies are summarized in Table 1.
Despite its wide usage, the systemic application of a scienceand risk-based strategy for the development of KFT methods has not yet been reported. The unmet need is augmented under current regulatory climate where quality by design (QbD) has started to play an increasingly central role. Successful applications of QbD principles to process development have been evidenced by publications as well as continued dialogue between the industry and regulatory authorities [23,24]. In parallel, applications to analytical method development has been progressed using the same science- and risk-based approach, including risk assessment and control strategy, to ensure that quality is built-in while methods are developed [23,25,26]. Most of the discussion, in the arena of QbD analytical methods, has centered on and will continue to evolve around strategies for adopting QbD principles in the development of robust analytical methodologies.
In recognition of the importance of the KFT method for water determinations and the unmet need of scientifically-driven and risk-justified KFT methods, this article proposes a comprehensive method development strategy (MDS) which is built upon a previously published MDS paper for RP-HPLC [25]. The strategy introduced here is consistent with QbD principles and the proposed position paper by PhRMA/EFPIA on QbD for analytical methodologies [27]. Three method examples were presented to illustrate relevant points. The first method was developed for elesclomol, which was used in a sterilized drug product. The second method was developed for pazopanib HCl, which was delivered by a solid oral tablet dosage form. The final method was developed for an API that contained weakly-bound water, known as a channel hydrate.
KFT Method Goal
Method intent including instrumentation capability of receiving laboratory, geographical location and access constraints to reagents must be evaluated before commencement of method development. As discussed in the introduction, water can have far reaching effects on the physical, chemical and biological properties of API and the performance of the drug product. Additional factors, such as patient safety, product efficacy and quality requirements, and knowledge of similar drug compounds, must also be considered.
While seemingly straightforward, setting the goal of a KFT method is convoluted with accuracy requirement for the assay method since water content is an input value for computing assay corrected on anhydrous and solvent free basis. Consequently, it becomes a critical factor limiting the drug content accuracy. Therefore it is necessary to control the KFT variability within the tolerable space of water content specification, and more so within the validated variability of the assay method. The criticality of KFT variability is also dictated by DP dosage form. For API intended for tablets or capsules, batch analysis data is the basis for setting specification if the API is a non-hydrate compound. Conversely, the stoichiometric hydrate value and batch data must be concurrently considered. More stringent water content specification is generally imposed for API intended for sterile formulations to ensure that water activity (aw) is below 0.6 to inhibit microbial activity in DP [9-11]. As such, the accuracy of the KFT method for low water levels will be a critical method performance attribute in the method design.
In summary, a multi-dimensional knowledge must be considered in the development of method goal, including (1) the type of water in the sample, (2) the targeted specification and regulatory expectation on similar drug compounds, (3) batch analysis results, (4) potential implications on safety and efficacy requirements, (5) quality compliance, and (6) end user limitations.
KFT Method Scouting and Evaluation
The first step in method scouting is to gather physiochemical properties of the sample, followed by evaluation of compatibility with KFT chemistries in light of extensively reviewed and published literature [2,28-30].
Understanding the Analyte
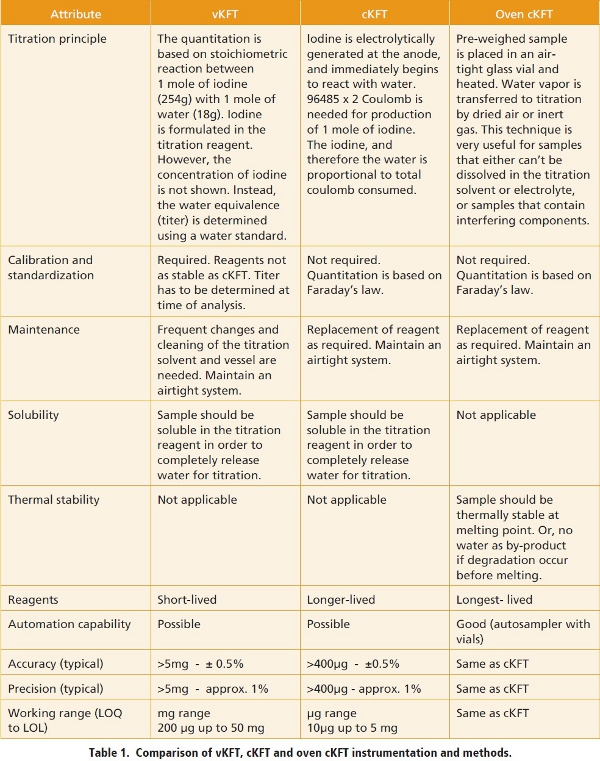
A variety of pharmaceutical compounds may present significant challenges to KFT due to potential interfering reactions. In cKFT, the API can potentially react with either iodine or iodide. In all KFT methods, the API can react with the reagents, such as the well-known reaction between aldehyde or ketone functionalities and methanol to release water as a by-product leading to positive bias. This can be avoided by use of methanol-free reagents [28,29]. API molecules that contain thiol can react with iodine, also resulting in positive error [29,30]. Other classes of compounds less known for compatibility issues include hydrazine derivatives, vinyl ethers, ascorbic acid, and aminoalcohols [28]. Direct oxidation by the generator electrode in cKFT is also a concern. Many organic compounds can be easily oxidized, such as anilines, phenols, and hydroquinones. Water determinations for such APIs should be conducted using either oven cKFT or vKFT to avoid side reactions that make the endpoint difficult to reach [2,28]. Beyond chemical reactivity, API molecules that are strongly acidic or basic can shift the pH out of its optimal range of 5 to 7, therefore use of buffers is required [2,28]. Finally, hygroscopic APIs and their hygroscopic behavior need to be carefully investigated because water present in API falls into two general categories [31-35]. The first category is liquid-phase water adsorbed to the surface of the API particles, commonly referred as surface water. The second category is water of hydration, which can either be tightly or loosely bound in the API crystalline lattice. Bound water can exist with a fixed stoichiometry [32,33] or as a variable hydrate [34,35]. Variable hydrates may equilibrate their moisture content with their environment over irreproducible timescales, it may be necessary to define a sampling and handling protocol involving rapid material transfers or humidity control. Thermal analysis, including DSC, TGA, and GVS, should be conducted with special care on hydrates to determine their sensitivity to environmental humidity as well as the minimal temperature needed to quantitatively release water from sample. Figure 1 shows the TGA thermogram of a channel hydrate API after being equilibrated at 33% RH to reach an approximate monohydrate. The channel water molecules, interacting with the API through hydrogen bonding, were released over a wide temperature range of 160 to 200°C. In this range, the material is fully dehydrated but has not yet begun to decompose.
Scouting and Evaluating KFT Methods
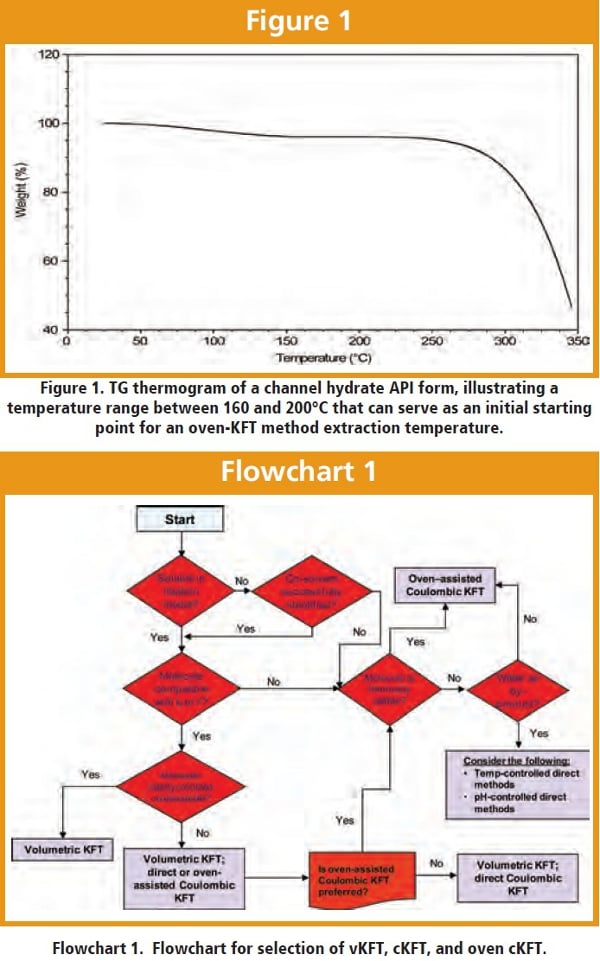
Flowchart 1 was developed to guide the scouting process in order to select the appropriate titration technique. Three major points to be considered include API solubility in the titration medium, compatibility with the KFT chemistry, and thermal stability. In cases where the choice of method is not clear from the information gathered, laboratory investigations should be conducted to illuminate some of the decision boxes shown in the flowchart.
Example 1 – Using LC/MS to investigate chemical compatibility of drug compound with titration chemistries
Elesclomol, a thiohydrazide derivative drug substance, was demonstrated to be a non-hygroscopic compound by GVS results. Water content for three batches at two stability conditions, as determined using direct cKFT, was displayed in Figure 2. The results for the same batch at the same stability condition varied from under 0.1% to over 1.4%w/w from one time interval to the next. The change in water content could not be correlated with the assay results. In fact, elesclomol content corrected on an anhydrous and solvent free basis had exceeded 102.0% (w/w) at multiple time points. No interfering side reactions between thiohydrazide and iodine was readily known since most sulfur containing compounds do not create problems except for mercaptans as previously discussed [30, 36]. Following the MDS approach, the titrated sample solution was pulled for LC and LC/MS investigation. The results, as illustrated in Figure 3, suggested that elesclomol was instantaneously oxidized by iodine to form the corresponding hydrazide degradation product, i.e. from –C(S)-N(CH3)-NH- to –C(O)-N(CH3)-NH-. This portion of iodine consumption was additive translating into an over-estimation of water content and causing high variability in the results. The LC/MS study helped conclude that direct cKFT was not suitable for elesclomol.

Example 2 – Using thermal analysis techniques to understand thermal stability and determine minimal temperature needed for oven cKFT
According to Flowchart 1, the next step in method selection was to evaluate the thermal stability of elesclomol in order to decide if it was amenable to oven cKFT analysis. Thermal analysis showed less than 0.2% weight loss from 30ºC to 150ºC, and onset of decomposition at around 181ºC. Consequently, the method was modified to use an oven accessory at 150ºC to liberate water from sample and avoid direct contact of elesclomol with iodine. The water content using the modified method was found to be 0.1%w/w or lower, in agreement with what observed in TGA thermograms.
KFT Method Selection and Risk Assessment
After the titration mode is decided, method variables should be systematically studied using structured risk assessment and experimental design tools.
Evaluating Method Performance and Understanding Critical Method Variables
Typical method variables include sample preparation, titration parameters, and oven parameters if relevant. These variables can be categorized as sample related, environmental dependent, or method and measurement related. Because of potential need to include a large number of variables in the robustness study, structured methodologies for risk assessment should be used to help assess their relative significance [37-39]. Example 3 illustrates the application of these tools to an oven cKFT method developed for pazopanib HCl.
Example 3 – Structured experimental design tools for evaluation of method robustness and ruggedness
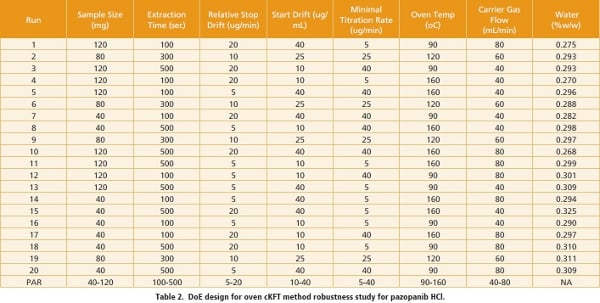
Based on the output of the risk assessment, a total of seven method parameters were recommended for a DoE study in order to understand their impact on method robustness. A 24 fractional factorial DoE design plus 4 center-points was selected, which resulted in a total of 20 titration runs. The results suggested that the method was robust within the ranges as summarized in Table 2. Water content from runs 2, 6, 9 and 19, in which the titration was conducted at the nominal method conditions, was 0.293, 0.288, 0.297, and 0.311% w/w, respectively.  The mean was 0.297% w/w with an RSD of 3.3%. Slightly larger variation was observed in the rest of the titration runs, with water content determined in the range from 0.268% to 0.325% with RSD of 4.9%. The proven acceptable ranges (PARs) for each of the seven method factors were therefore determined and summarized in Table 2. Operating the method within the PARs will pose little risk to the registered specification of NMT 1.0% w/w. The conclusion was supported by other statistical results from the DoE study, including the half-normal plot as displayed in Figure 4.
The mean was 0.297% w/w with an RSD of 3.3%. Slightly larger variation was observed in the rest of the titration runs, with water content determined in the range from 0.268% to 0.325% with RSD of 4.9%. The proven acceptable ranges (PARs) for each of the seven method factors were therefore determined and summarized in Table 2. Operating the method within the PARs will pose little risk to the registered specification of NMT 1.0% w/w. The conclusion was supported by other statistical results from the DoE study, including the half-normal plot as displayed in Figure 4.
Method ruggedness, based on a measurement system analysis (MSA) design in Figure 5, was conducted to evaluate noise factors including variability in analysts, instruments, reagents, and laboratories. Study results indicated that the most significant variability was associated with sample preparation. In contrast, the variability resulted from the confounded factors of “analyst/day/ instrument” was only a quarter of the magnitude of that caused by sample preparation (Figure 6 and Table 3). Statistical multi-parameter DoE provides not only effective experimental designs but also allows result assessment that is not biased towards any parameters. In some cases, however, direct examination of a suspected critical parameter can be used to either avoid complex experimental protocols or to understand parameter impact in an extended range. For example, for the variable hydrate API discussed previously, the sensitivity of the water content to temperature for a single batch can be quickly charted beyond a typical DoE range to identify the most robust temperature (Figure 7). The results, when extended to multiple batches, suggested that temperatures of below 150oC are not robust; instead higher temperatures such as from 170 to 200oC would more likely yield robust results and avoid potential temperature calibration or setting issues with the instrument.

KFT Method Control Strategy
An appropriate system suitability test, depending on the expected water level in the sample, may be the only control element needed in ensuring performance of the selected method. This is true for the method discussed in Example 3, for which the PARs for all seven method parameters were wider than their corresponding performance ranges verified during routine maintenance.  Naturally, the acceptable control strategy for this method will simply be good laboratory practices plus an appropriate system suitability test. An example of system suitability test for direct titration cKFT is included in Flowchart 2.
Naturally, the acceptable control strategy for this method will simply be good laboratory practices plus an appropriate system suitability test. An example of system suitability test for direct titration cKFT is included in Flowchart 2.
Occasionally, identification of a backup KFT method may become part of the control strategy. In cases where vKFT and cKFT can be used interchangeably, or if the sample can be handled by either direct titration or thru oven, an alternative KFT method can be selected as the backup. Statistical evaluation of methods equivalency should be conducted to qualify the backup method. In the case that no other KFT mode can be used as the backup method, water content results from a range of batches can be verified using a thermal method such as LOD or TGA in order to detect potential systematic bias. It should be noted, however, that neither LOD nor TGA is specific for water determination, and correction for other volatiles should be included during data evaluation. Example 4 illustrates an approach for KFT equivalency study.
Example 4 – Method equivalency study in evaluation of a backup method
In order to demonstrate whether direct and oven cKFT methods can be used interchangeably for water determination in pazopanib HCl, a method equivalency study was performed. An equivalence assessment was conducted against the acceptance criterion (ø) of 0.2% absolute, which was derived from 20% relative of the registered specification limit of NMT 1.0%w/w. The acceptance criterion (ø) is the largest difference in mean water content that is considered not to cause practical significance in the testing result. Ten batches of API were selected to present a suitable challenge to the methods being assessed. 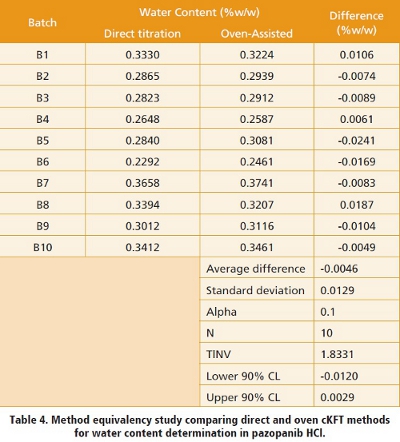 Ten pairs of water content values were generated and used for data comparison and statistical analysis. Based on the data gathered from the study, a 90% confidence interval for the mean was achieved with paired differences well within the upper and lower allowable limits of +/- 0.20%w/w, with lower and higher 90% confidence interval of – 0.012% and 0.0029%, respectively (Table 4). The statistical evaluation suggests the two methods are equivalent, and supports the conclusion that the method contains no systematic bias that could affect the reported water content values.
Ten pairs of water content values were generated and used for data comparison and statistical analysis. Based on the data gathered from the study, a 90% confidence interval for the mean was achieved with paired differences well within the upper and lower allowable limits of +/- 0.20%w/w, with lower and higher 90% confidence interval of – 0.012% and 0.0029%, respectively (Table 4). The statistical evaluation suggests the two methods are equivalent, and supports the conclusion that the method contains no systematic bias that could affect the reported water content values.
Final Validation for Further Verification of KFT Method Performance
To this point, the discussion has focused on obtaining integrated knowledge of sample characteristics together with titration chemistries, and risk understanding, control and management (Flowchart 3). In contrast to conventional approaches for method development, method risks are identified and understood during method evaluation process, and are thoroughly investigated during the robustness and ruggedness studies. 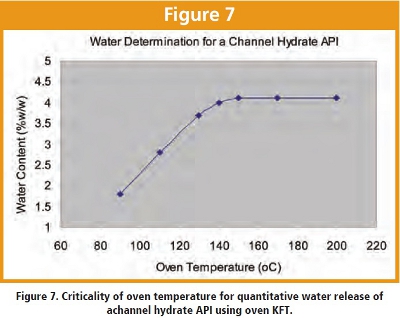 The systemic approach as described ensures high chance of success for accurate and robust KFT method. Final method validation is conducted for the purpose of further verification of method performance characteristics that is built into the method during the development process, as well as for enhanced knowledge of the method.
The systemic approach as described ensures high chance of success for accurate and robust KFT method. Final method validation is conducted for the purpose of further verification of method performance characteristics that is built into the method during the development process, as well as for enhanced knowledge of the method.
The method performance attributes recommended for final method validation are summarized in Table 5, including method accuracy, linearity and precision. The accuracy is evaluated by spiking a known amount of pure water or a solid hydrate standard into the sample, and is calculated by recovery. Precision is evaluated to allow for assessment of method intermediate precision as described in Example 3. By varying sample quantity, linearity can be conveniently verified. The results from this finalization step, together with learning from method evaluation and selection steps, constitute the integrated knowledge space for the KFT method, and serve as the baseline for continuous monitoring and potential improvement of the method after its implementation.

Conclusion
A systemic KFT method development strategy has been proposed, which emphasizes on detailed understanding of chemical compatibility between the sample and KFT reagent as well as the electrochemical reactivity of the sample. Enabling tools such as the Fishbone diagram and FMEA for risk assessment, DoE for robustness study, and MSA for method ruggedness evaluation are fundamental to a structured investigation and understanding of the impact of method variables and parameters. Critical method parameters and factors are identified after evaluating all experimental results against the measurement requirements, which are defined in the method goal. Critical parameters are either monitored or controlled in the method to mitigate the risk, or eliminated by developing a new method.
Several examples of the application of KFT MDS at various stages have been given to illustrate the working principles. Multiple flowcharts have been presented to guide the evaluation and selection process for appropriate KFT methods. Since success in developing a robust KFT method primarily depends on the physiochemical and thermal compatibility of API, an example was provided to highlight significant risks in a KFT method where the chemistry between the sample and the titration was not thoroughly understood. Other examples have demonstrated that operating ranges for typical titration parameters, including start drift, stop drift, titration rate, and oven temperature, are robust over a wide range so that their controls can be maintained through regular instrument services. Finally, our studies have suggested that oven cKFT is a rugged method for water determinations for many classes of API molecules; together with sampling automation, oven cKFT may offer an advantage over direct KFT methods for many projects in the future.
Acknowledgements
The authors would like to thank Ms. Anna Caltabiano and Dr. Lianming Wu (Chemical Development, GSK) for their help in generating chemical compatibility data, and Ms. Rachel Forcino (Pharmaceutical Development, GSK) for her help in providing thermal analysis data, and Mr. Uday Bhatt (Pharmaceutical Development, GSK) for participating in the MSA study. The authors also thank Dr. Jianhua Shen (Synta Pharmaceuticals) for providing water content data that was used for construction of Figure 2. The authors would also like to acknowledge Mr. Alberto G. Garofalo, Mr. Todd W. Koretke, Mr. Jose M. Fueyo, Mr. Liang-chi Li, and Mr. Richard P. Jones (Pharmaceutical and Chemical Development, GSK) for their contributions to Table 5.
References
1. ICH Q6A: Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances.
2. MacLeod, S. K., “Moisture determination using Karl Fischer titrations,” Anal. Chem., 1991, 63, 557A-566A.
3. Connors, K. A., “The Karl Fischer titration of water,” Drug Dev. Ind. Pharm., 1988, 14, 1891-1903.
4. Cachet, T., Hoogmartens, J., “The determination of water in erythromycin by Karl Fischer titration,” J. Pharm. Biomed. Anal.,1988, 6(5), 461-72.
5. Lindquist, J., “Determination of water in penicillins using fast Karl Fischer reagents and electronic end-point optimization,” J. Pharm. Biomed. Anal., 1984, 2, 37-44.
6. Yoshioka, S., Stella, V.J., “Stability of Drugs and Dosage Forms”, Springer, 2000.
7. Constantino, H.R., Langer, R. and Klibanov, A.M., “Aggregation of a lyophilized pharmaceutical proteins, recombinant human albumin: Effect of moisture and stabilization by excipients”, Biotechnology, 1995, 13, 493-496.
8. Constantino, H.R., Langer, R. and Klibanov, A.M., “Moisture-induced aggregation of lyophilized insulin”, Pharm Res., 1994, 11, 21-29.
9. Troller J.A., Bernard D.T., and Scott V.W., “Measurement of water activity. In: Compendium of Methods for the Microbiological Examination of Foods”, American Public Health Association, Washington, DC, 1984, 124-134.
10. Warner A.W., Stafford J.D., and Warburton R.E., “Establishing a microbial control strategy in active pharmaceutical ingredients,” American Laboratory 2007, 39, 15-18.
11. “Microbiological attributes of non-sterile pharmaceutical products application of water activity determination,” Pharmacopeial Forum, 2002, 28, 2009-2013.
12. Fischer, K., “Neues Verfahren zur maßanalytischen Bestimmung des Wassergehaltes von Flüssigkeiten und festen Körpern,” Angewandte Chemie, 1935, 48, 394–396.
13. Schlink, R., Dengler, C., Kirschenbuhler, P., Surborg, K., “Automatic Karl Fischer water determination in drugs,” GIT Labor-Fachzeitschrift, 2003, 47, 141-145.
14. Chen, Q., and Wang, Y.W., “Challenges in the Determination of Water Content by Karl Fischer Titration During In-Process Control,” Pharm. Rev. 2009, 12, 48-54.
15. Kestens, V.; Conneely, P.; Bernreuther, A. “Vaporisation coulometric Karl Fischer titration: A perfect tool for water content determination of difficult matrix reference materials,” Food Chem. 2008, 106, 1454–1459.
16. Adam M., Dobiš, Bajerová P., Ventura K., “Comparison of various methods for determination of water in white yoghurts”, Food Chemistry, 2009, 115, 1069-1073.
17. Spahn, G., Plettenberg, H., Nagel, H., Kahl, E., Klinger, H.M., Günther, M., Mückley, T., Hofmann, G.O. “Karl Fischer titration and coulometry for measurement of water content in small cartilage specimen”, Biomedizinische Technik 2006, 51, 355-359.
18. Margolis, S. A.; Vaishnav, K.; Sieber, J. R. “Measurement of water by oven evaporation using a novel oven design – Part 1: Water in watersaturated 1-octanol, coal, cement, and refined oils.” Anal. Bioanal. Chem. 2004, 380, 556–562.
19. Margolis, S. A.; Vaishnav, K.; Sieber, J. R. “Measurement of water by oven evaporation using a novel oven design – Part 2: Water in motor oils and motor oil additives.” Anal. Bioanal. Chem. 2004, 380, 843–852.
20. Bernreuther, A.; Klein, C. L. “Water content determination of the candidate reference material for gliadin from European wheat (IRMM- 480).” In M. Stern (Ed.), Proceedings of the 19th meeting Working Group on prolamin analysis and toxicity, Zwickau, Germany: Verlag Wissenschaftliche Scripten, 2005, pp. 99–108.
21. Bruttel, P., Schlink, R., “Water Determination by Karl Fischer Titration,” Metrohm Ltd, Herisan, Switzerland, 2006.
22. Scholz, E., “Fifty years of Karl Fischer titration,” CLB, Chemie fuer Labor und Betrieb, 1985, 36, 381-3.
23. Yu, L., “Pharmaceutical quality by design: product and process development, understanding and control,” Pharm. Res. 2008, 25(4), 781-791.
24. Gavin, P.F., Olsen, B.A., “A quality by design approach to impurity method development for atomoxetine hydrochloride (LY139603),” J. Pharm. Biomed. Anal., 2008, 46, 431-441.
25. Li, Y.; Terfloth, G., Kord, A. S. “A systematic approach to RP-HPLC Method Development in a pharmaceutical QbD environment,” Am. Pharm. Rev., 2009, 12, 87-95.
26. Borman, P., Nethercote, P., Chatfield, M., Thompson, D., Truman, K., “The application of quality by design to analytical methods,” Pharm. Tech., Oct., 2007
27. Schweitzer, M., Pohl, M., Hanna-Brown, M., Nethercote, P., Borman, P., Hansen, G., Smith, K., Larew, J., “Implication and opportunity of applying the principles of quality by design to analytical measurements,” Pharm. Tech. Europe, 22 (2), 2010.
28. Mitsubishi Chemical Corporation, Karl Fischer Reagents Technical Manual, 2000.
29. Stern M., “Reagent Selection in the Karl Fischer Analysis of Pharmaceutical Products,” American Laboratory, 2006, 38, 28-29.
30. Sherman, F.; Kuselman, I. “Water determination in drugs containing thiols,” Int. J. Pharm. 1999, 190, 193–196.
31. Byrn, S. R.; Pfeiffer, R. R.; Stowell, J. G. Solid-state chemistry of drugs. SSCI: West Lafayette, IN, 1999.
32. Datta, S., and Grant, D.J.W., “Crystal structures of drugs: advances in determination, prediction and engineering,” Nature Reviews Drug Discovery, 2004, 3, 42-57.
33. Vogt, F. G.; Brum, J.; Katrincic, L. M.; Flach, A.; Socha, J. M.; Goodman, R. M.; Haltiwanger, R. C. “Physical, crystallographic, and spectroscopic characterization of a crystalline pharmaceutical hydrate: understanding the role of water,” Cryst. Growth & Des., 2006, 6, 2333-2354.
34. Vogt, F. G.; Dell’Orco, P. C.; Diederich, A. M.; Su, Q. ; Wood, J. L.; Zuber, G. E.; Katrincic, L. M.; Mueller, R. L.; Busby, D. J.; DeBrosse, C. W. “A study of variable hydration states in topotecan hydrochloride,” J. Pharm. Biomed. Anal., 2006, 40, 1080-1088.
35. Morris, K. and Rodriguez-Hornedo, N., Hydrates. In: J. Swarbrick and J.C. Boylan, Editors, Encyclopaedia of Pharmaceutical Technology 7, Marcel Dekker, New York, 1993, 393–440.
36. “Hydranal – Manual, Eugen Scholz Reagents for Karl Fischer Titration,” Riedel-de-Haen, Inc. 1997, pg. 97.
37. Kumamoto, H., Henley, E., Probabilistic Risk Assessment and Management for Engineers and Scientists, 2nd ed., IEEE Press., Piscataway, NJ, 1996.
38. Scipioni, A., Saccarola, G., Centazzo, A., Arena, F., “FMEA methodology design, implementation and integration with HACCP system in a food company,” Food Control, 2002, 13, 495-501.
39. Ellison, S. L. R.; Barwick, V. J. “Using validation data for ISO measurement uncertainty estimation - Part 1. Principles of an approach using cause and effect analysis” Analyst, 1998, 123, 1387-1392.
Leon Zhou, Ph.D.,Team Manager in the Chemical Development Analytical Sciences department at GlaxoSmithKline. He has over fifteen years of experience in analytical development with research interests spanning from chromatography to spectroscopy methodologies. He currently leads a team of analytical scientists focused on assets in late phase development.
Jerome M. Socha, Scientist in the spectroscopy and compendial methods group within the Chemical Development Analytical Sciences department at GlaxoSmithKline. His interests include compendial methods, classical analytical methods, and atomic spectroscopy. He has more than 20 years of experience in the application of Karl Fischer titration to drug substances and other materials using direct and oven methods.
Frederick G. Vogt, Ph.D., Team Manager of spectroscopy and compendial methods group within the Chemical Development Analytical Sciences department at GlaxoSmithKline. His interests include techniques such as NMR, x-ray diffraction, and vibrational spectroscopy, as applied to drug substance crystallization and form control, drug product and excipient characterization and secondary manufacturing processes, and patent support.
Sarah Chen, Ph.D., Investigator in the Chemical Development Analytical Sciences Department at GlaxoSmithKline. She has over ten years of experience in analytical development for process research for early phase and late phase projects. Her interests include stationary phase and separation mechanism, analytical method development and validation, trouble shooting and problem solving.
Alireza Kord, Ph.D., Director, heads the Analytical Sciences Department in Chemical Development at GlaxoSmithKline, King of Prussia, PA. He has close to twenty years of experience in the pharmaceutical industry and assumed his current position in 2003. His educational background and scientific interest is in the field of separation sciences including HPLC, CE, GC, etc.