Introduction
Protein products are commonly dried due to the inadequate stability of proteins in aqueous solution. When a given protein formulation is dried using different drying methods, such as lyophilization, spray drying, or vacuum drying, the resulting products can exhibit large differences in the physico-chemical properties of the solids such as specific surface area (SSA), surface composition and molecular mobility etc [1]. Variation in these physico-chemical properties can result in significant differences in both in-process and storage stability [2]. For example, storage stability of a rhuMAb was found to be better in freeze-dried formulations than spray-dried product, even though no differences were observed in the secondary structure of the protein prepared by both techniques [3]. It is well known that powders produced by spray drying usually have a higher SSA than corresponding product prepared by lyophilization. Moreover, due to some surfactant character, more protein may be distributed near the surface of the particle in spray-dried product. These differences in surface properties may explain the better stability of rhuMAb in the lyophilized product than in the spray-dried sample. The importance of surface properties in impacting stability of proteins has been reviewed recently, and critical factors affecting the drying method selection were summarized [2].
Whether different drying methods are used to prepare the same formulation or a given method is employed where processing conditions during drying are modified, the net result is a change of the thermal history. Since thermal history plays an important role in determining the physico-chemical properties and long-term stability of the product, a better understanding of the role of thermal history will assist in design of appropriate drying process that will improve product quality [3]. In this paper, we focus on the freeze drying process since this process is, by far, the most commonly used drying process for protein products [4,5]. We aim to examine the challenges and opportunities in maximizing protein stability through lyophilization process optimization. Since both protein native structure and molecular mobility in the dried product play an important role in the protein stability, it is necessary to explore the impact of thermal history on the global, local molecular mobility and protein structure. In the next section, the impact of thermal treatment on product stability is discussed during the freezing, primary drying and secondary drying stages, respectively. Then, the design of appropriate thermal treatment process to improve protein stability is proposed. Finally, a brief summary is provided at the end.
Annealing in the Frozen State During Step
The freezing step is the first step in the freeze drying process, and the objective of this step is to convert most of the water to ice and thus separate ice from the solutes. Generally, a low shelf temperature (~−40°C or −50°C) is used, and the product is typically kept at this temperature for several hours to ensure that all solution has transformed into solid. As freezing proceeds beyond the first nucleation and formation of ice, the solute phase becomes highly concentrated and is termed the “freeze concentrate.” A process modification in the freezing step is called annealing, which involves holding the product at a temperature above the final freezing temperature for a defined period [6]. The annealing temperature should be between the ice melting temperature and the glass transition temperature of the freeze concentrate (Tg’) and holding times are normally several hours [6,7].
One purpose of annealing is to completely crystallize bulking agents such as glycine or mannitol in the formulation during the freezing stage [6, 7]. A crystalline matrix provides a framework or support to prevent the cake collapse even if the amorphous phase (i.e., drug, buffer, and stabilizer) is being dried above its collapse temperature. In such cases, it is necessary to have a product with relatively low dose so that one may add a stabilizer at a weight ratio in excess of 1:1 with the drug (plus buffer) and yet add a bulking agent well in excess of the amorphous phase (roughly 3:1 bulking agent to amorphous phase) without producing a total solute concentration much greater than 10%. The advantage of a formulation containing a stabilizer plus a bulking agent is that, in such cases, the formulation can be easily freeze dried at temperatures well in excess of the collapse temperature of the amorphous phase, resulting in a much shorter freeze-drying cycle, and yet obtain an elegant product. Since the bulking agent is not expected to play a role in stabilization, the drug storage stability is normally close to that of a system freeze dried without the crystalline bulking agent. However, failure to crystallize the bulking agent has the potential of depressing the Tg’, particularly with glycine, or the Tg, more serious with mannitol, and potentially compromising storage stability by crystallizing from the solid during storage and releasing the water contained in the amorphous bulking agent into the drug phase. In addition, in many systems, annealing will also reduce the resistance to water vapor transport in the porous cake, and will therefore, accelerate primary drying. For a pure amorphous system like sucrose, annealing will allow smaller ice crystals to melt and larger ones to grow according to the Ostwald ripening mechanism [6,7]. Thus a larger pore size is obtained after the freezing process, which results in less flow resistance and a shorter primary drying cycle. In addition annealing decreases the vial-to-vial drying variations and thus results in a more homogeneous batch [6,7].
Annealing above the Tg’ during freezing may be quite useful; however, mobility would be enhanced in the system, and potential drug degradation or modification of protein structure during the annealing could predispose the formulation to greater instability during storage. The documented effects of annealing in the frozen state on protein stability are variable. For example, it was reported that annealing accelerates phase separation in the freeze-concentrated liquid phase, and thus causes a conformational change in hemoglobin [8]. Annealing was also found to cause an increase in the level of aggregation of Interlukin-6 relative to an untreated control, both during lyophilization and during storage [9]. On the other hand, Sarciaux and coworkers observed that annealing resulted in significantly less aggregation of bovine IgG in the dried state, with a corresponding decrease in SSA of the annealed system [10]. Webb etc investigated the effect of annealing on stability of recombinant human interferon-g (rhIFN-g) during lyophilization and spray-lyophilization [11]. Figure 1 shows the FTIR structure of rhIFN-g in different formulations with and without an annealing step. Upon annealing, the intensity of the major α-helix band at 1656 cm-1 increases significantly and also the band narrows for all formulations. The more native-like protein secondary structure is consistent with the observed decrease in protein aggregation level [11]. A recent study of an IgG1 monoclonal antibody in sucrose formulations showed that annealing did not result in significant change in either protein secondary structure or storage stability [1]. Although one may expect that annealing above Tg’ will lead to protein unfolding due to higher mobility above Tg’, recent work showed that protein unfolding was negligible in freeze concentrated systems on the time scale of annealing process even well above Tg’ [12]. Calculations showed that even at 20°C above Tg’, the estimated half-life for protein unfolding for the two proteins studied was on the time scale of years due to a strong coupling between system viscosity and unfolding rate, especially at high stabilizer level. Also the reduction in specific surface area of the freeze-dried product upon annealing may stabilize protein due to a lower protein surface accumulation [2, 11]. These kinetics and surface factors may explain the beneficial effect observed on some proteins upon annealing in frozen solution. Thus, even though one might theoretically expect instability during annealing the frozen system, and instability has been documented in some systems, stability is often not impacted and on occasion might even be improved. Clearly, one must investigate each new case in order to utilize the potential benefi t of annealing process!

Drying Above Tg’ during Primary Drying
The purpose of primary drying is to remove ice by sublimation under very low pressure such as 100 mTorr (13.3 Pa). The shelf temperature needs to be raised to supply the heat removed by ice sublimation. Typically, the sample temperature should be lower than the collapse temperature (Tc), otherwise the freeze-dried product loses macroscopic structure and collapses during freeze drying [13]. The target product temperature is generally 2 to 5°C below the Tc or Tg’ to get an elegant cake and a short drying cycle. However, as noted earlier, when a crystalline bulking agent is included in the formulation, macroscopic collapse may not occur even the product is dried above Tg’ [13]. In this case, amorphous materials may undergo viscous flow on a microscopic scale, collapsing around crystalline material, but not exhibiting macroscopic collapse.
The impact of conducting primary drying above Tg’ on protein stability has been investigated with mixed results. Lueckel and co-workers reported that structural collapse of a lyophilized cake results in increased levels of aggregation of IL-6 in a glycine/sucrose formulation [9]. Jiang and coworkers [14] have investigated the effect of collapse on the biological activity of freeze-dried catalase, β-galactosidase, and lactate dehydrogenase (LDH), and it was found that the resulting activity loss for catalase and β-galactosidase was small, but the activity loss for LDH was rather significant. The authors suggested that the large loss of activity for LDH may be due to partial melting of the samples under the drying conditions used. Wang and co-workers also studied the effect of process variation on long-term stability of Recombinant Factor VIII (rFVIII) and α-amylase in sucrose/glycine formulation [15]. Using three different drying cycles, normal and collapsed cake structures were obtained. The stability profiles for three different cycles are shown in Figure 2. For rFVIII, there is no difference in the biological activity for product freeze-dried under different drying conditions when stored at 5 or 25°C. At 40°C storage, however, the stability of the collapsed product (Cycle 3) appeared to be even better than that of normal product freeze-dried with a conservative cycle (see Figure 2). For α-amylase, there was no impact of process temperature on stability at any of the three storage temperatures. Suryanarayanan et al investigated the recovery of LDH activity immediately after freeze drying with two different cycles in glycine-raffinose or glycinetrehalose system [16]. There was no significant loss in LDH activity in the collapsed product, and the same conclusion was made by Luthra and co-workers in studies on LDH using a humidity controlled mini freeze dryer [17]. Four different formulations (no lyoprotectant, 5% w/v sucrose, trehalose or raffinose in citrate buffer and 0.05% w/v Tween 80) were investigated, and structural collapse was found to have no negative impact on LDH activity recovery after primarydrying, even in the formulation without any lyoprotectant. Colandene et al investigated the FTIR structure and long-term stability for a monoclonal antibody in a formulation containing 8% sucrose and high levels of protein [18]. When primary drying cycles were performed at product temperatures above Tg’, no apparent collapse was noticed. The authors suggest that the presence of high protein concentration significantly increased the collapse temperature, and prevented the macroscopic collapse even without a crystalline bulking agent. FTIR study showed that aggressive and conservative cycles induced the same minor secondary structural changes in the protein structure relative to the liquid state. SEC-HPLC analysis indicated that there was no difference in the physical stability upon storage at 5, 25 and 40°C for up to 12 months. Thus, there are numerous examples of freeze drying above Tg’ without producing any significant negative impact on product stability, either during the process or during subsequent storage. However, Passot and co-workers studied the long-term stability of lyophilized toxin A and B proteins in two formulations containing either Polyvinylpyrrolidone (PVP) or mannitol as bulking agent [19]. After 6 months of storage at both 4 and 25°C, the antigenic activity of the toxins in both formulations was lower for those processes where the primary drying was performed with a product temperature higher than Tg’. Recently, it was reported that while collapse during primary drying (using an aggressive cycle) did not have a significant effect on either the in-process or storage stability of an IgG1 in a sucrose formulation [20]. Collapse during storage, however, did have a distinct negative impact on protein stability.
It is important to distinguish between collapse as a cosmetic defect versus a product quality attribute that could result in sub-therapeutic dosing or toxicology issues from a degradation product. Given the above findings regarding the impact of collapse on protein activity, it seems that there is no general rule for predicting the impact of drying above Tg’ on stability of proteins. The impact of collapse may depend on the sensitivity of each protein, the stabilizer/bulking agent in the formulation and the thermal history in the drying cycle, and thus needs to be established on a case-by-case basis. Since collapse may not necessarily be detrimental to the long-term stability of freeze-dried proteins, it is possible to perform primary dry at temperature above the Tg’ by judiciously selecting the formulations. In order to provide a very robust formulation for freeze drying, a formulation combining crystalline and amorphous components may be used. Such a robust formulation is much less sensitive to temperature variations, and they can be dried at higher temperatures without macroscopic product collapse or loss of any product quality attribute.
Thermal Treatment of a Dried Glass During Secondary Drying
 Immediately after primary drying, an amorphous product still contains 15-25% water on a dried solids basis, depending on the formulation [13]. The major objective of secondary drying is to reduce the residual moisture content to a level optimal for stability, which is usually less than about 1%. It was found that the water desorption rate at a given drying temperature decreases dramatically in the first 1-2 hours, followed by a period of much lower drying rate [21]. The residual water content appears to approach a plateau level after drying at a given temperature for about 4-6 hours, and drying for longer times does not significantly reduce the moisture content. The water desorption rate is highly dependent on drying temperature [13], and it is generally better to run at a high shelf temperature (and therefore, high product temperature) for a short time than a low temperature for a long period. Depending on the formulation, the secondary drying temperature can vary in the range of 40 to over 50°C, even for proteins.
Immediately after primary drying, an amorphous product still contains 15-25% water on a dried solids basis, depending on the formulation [13]. The major objective of secondary drying is to reduce the residual moisture content to a level optimal for stability, which is usually less than about 1%. It was found that the water desorption rate at a given drying temperature decreases dramatically in the first 1-2 hours, followed by a period of much lower drying rate [21]. The residual water content appears to approach a plateau level after drying at a given temperature for about 4-6 hours, and drying for longer times does not significantly reduce the moisture content. The water desorption rate is highly dependent on drying temperature [13], and it is generally better to run at a high shelf temperature (and therefore, high product temperature) for a short time than a low temperature for a long period. Depending on the formulation, the secondary drying temperature can vary in the range of 40 to over 50°C, even for proteins.
Traditional freeze drying practice has often used low secondary drying temperature (~ 25°C) for proteins, presumably in the belief that the protein would denature at higher temperature. However, it is found that dry protein powders are extremely stable to thermal denaturation. In contrast to the low denaturation temperatures (about 50 to 80°C) in protein aqueous solution, the denaturation temperature (Td) in dry glass (~1% moisture) is found to be over 150°C for human growth hormone (hGH), an IgG1 antibody, human serum albumin [22, 23]. Even in the early stage of the secondary drying process, the denaturation temperature in the relatively dry (<15% water) solid state is well over 100°C for hGH [24]. Thus protein denaturation during secondary drying process is not an issue at drying temperatures well below about 100°C. 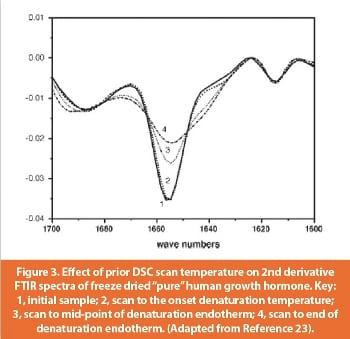
The impact of thermal history on protein denaturation in freeze dried formulations has been recently studied with hGH as a model [23]. Upon scanning to different temperatures around Td, the sample was cooled to room temperature and protein stability was analyzed with FTIR, HPLC and DSC. Figure 3 shows the impact of prior DSC scan temperature on the 2nd derivative FTIR spectra of freeze dried pure hGH. Curve 1 represents the initial freshly freeze dried material, which shows the characteristic a-helix band at 1658 cm-1. Curve 2 is the spectra of a sample scanned to the onset of denaturation (~150°C) and then cooled. The difference between curve 1 and 2 is not significant; suggesting that prior scan to the onset of denaturation did not cause any obvious damage to secondary structure. The α-helix band intensity is largely reduced in curve 3 and 4 where the sample has been scanned to the midpoint and endpoint of Td, respectively. Thus, the protein was unfolded upon scanning into the denaturation endotherm region in curve 3 and 4. HPLC and DSC results also indicated that brief exposure of the protein to temperatures slightly less than the onset Td did not cause significant damage to the protein [23]. These results suggest that thermal denaturation of protein in solid state occurs only well above the Tg and drying at a high temperature well below Tg will not cause protein unfolding in the dry solid.
A high drying temperature during secondary drying not only results in fast desorption of water and thus a short drying cycle, it also has other important consequences for product quality. Thermal treatment of a glass below its Tg for a short period of time was reported to improve the chemical stability of some pharmaceuticals, such as moxalactam and aspartame in amorphous solids [25, 26]. Thus, better stability of a pharmaceutical may be achieved by performing secondary drying at a higher temperature or using heat treatment post-drying in an oven. This thermal treatment process, carried out on a dried glass, is also called physical aging, structural relaxation or annealing [27]. It should be pointed out that while this physical aging process is often called annealing in the literature, this “annealing” is totally different from the annealing process during the freezing stage we discussed earlier. First, the aging process here is performed on a dried glass, below the system Tg and thus is different from the annealing process in a frozen solution which is performed above the system Tg’. Second, the main purpose of aging process is to facilitate the structural relaxation in a non-equilibrium glass, which is also different from the crystallization of crystalline bulking agent for the annealing process in the freezing step. Since the physical aging process may be optimized to maximize product quality, it is of great importance to understand how thermal treatment will impact the molecular mobility and the stability of pharmaceuticals in a dried solid.
Annealing Effect on Global Mobility
 A glass will relax toward the ‘‘equilibrium glassy state’’ spontaneously due to its high free energy, and many properties continue to change during this process. For example, volume decreases, structural order increases (i.e., configurational entropy decreases) and heat is released (enthalpy relaxation) [28]. Physical aging studies on polymers and small molecule glasses have shown that the structural relaxation time increases during annealing (i.e. global mobility decreases) [27, 28]. Figure 4 shows the calculated effect of aging temperature on the structural relaxation time in a sucrose glass. The mean structural relaxation time increases toward the equilibrium curve with increasing annealing temperature [29]. There is growing experimental evidence for a coupling between pharmaceutical stability and molecular mobility in amorphous solids, and thus a system with lower mobility normally shows better stability [30, 31]. Since aging causes a decrease in global mobility, one may expect that annealing below Tg would stabilize a glass. The possible beneficial effect of annealing was recently demonstrated with Maillard reaction in a glassy system of lysine in trehalose, sucrose, and glucose [32]. Aging was found to lower the reaction rate by about 20%. It was suggested that annealed glass becomes denser which may slow down the rate of diffusion of the reactants and thus improves the stability.
A glass will relax toward the ‘‘equilibrium glassy state’’ spontaneously due to its high free energy, and many properties continue to change during this process. For example, volume decreases, structural order increases (i.e., configurational entropy decreases) and heat is released (enthalpy relaxation) [28]. Physical aging studies on polymers and small molecule glasses have shown that the structural relaxation time increases during annealing (i.e. global mobility decreases) [27, 28]. Figure 4 shows the calculated effect of aging temperature on the structural relaxation time in a sucrose glass. The mean structural relaxation time increases toward the equilibrium curve with increasing annealing temperature [29]. There is growing experimental evidence for a coupling between pharmaceutical stability and molecular mobility in amorphous solids, and thus a system with lower mobility normally shows better stability [30, 31]. Since aging causes a decrease in global mobility, one may expect that annealing below Tg would stabilize a glass. The possible beneficial effect of annealing was recently demonstrated with Maillard reaction in a glassy system of lysine in trehalose, sucrose, and glucose [32]. Aging was found to lower the reaction rate by about 20%. It was suggested that annealed glass becomes denser which may slow down the rate of diffusion of the reactants and thus improves the stability.
There are only a few examples of stabilization of amorphous pharmaceuticals by high temperature treatment. The first report documented the increased stability (about 20%) of moxalactam disodium formulated with 12% w/w mannitol if secondary drying were carried out at 60°C rather than 40°C; even though the moisture contents were essentially identical [33]. A more extensive study was later carried out on the same system, and thermal treatment was performed post-lyophilization in a lab oven [25]. It was found that as annealing temperature and time increased, the global mobility (measured with τβ) decreased. Figure 5 shows the time-dependent degradation upon storage of fresh and annealed samples. Note that the degradation rate decreases upon annealing, and there is a systematic improvement in long-term stability for the thermal treated samples. Even though the initial degradation was a little higher due to the thermal treatment, annealed samples actually exhibited less degradation at the end of the storage period. The annealing effect on a dipeptide system was also investigated, and efforts have been made to study the relationship between thermal treatment, molecular mobility and chemical stability [26]. Chemical assay showed that the rate of degradation via cyclization in aspartame/disaccharide glass decreases significantly upon annealing. Similarly, degradation of sodium ethacrynate (ECA) via dimerization reaction was also studied [34]. Annealing ECA in amorphous solids improved chemical stability, as shown by the decrease in degradation rate constant relative to the control. It was also found that thermal treatment slowed down the global motions, as expected. 
Annealing Effect on Local Mobility
Solid-state NMR can be used to study local rotational motions with the timescale between microseconds and seconds. The effect of annealing on local mobility in aspartame/sucrose systems was investigated using solid-state 13C NMR, and it was found that the NMR measured mobility decreased slightly upon annealing [35]. However, there was no significant annealing impact on local mobility in the corresponding aspartame/trehalose formulation [35]. In another NMR study, the T1ρ relaxation time in starch was measured as a function of aging time at different humidity levels [36]. It was found that as starch annealed at temperature below Tg and at 30% R.H., T1ρ increased and local mobility decreased. While these studies showed that physical aging may slow the local motion of molecule [35, 36], other studies show that physical aging does not significantly impact the beta relaxation of polymers like PMMA [37] , glass formers such as silica, glycerol and xylitol [38] and ECA/sugar samples [34]. Thus, the impact of physical aging on the local mobility (beta relaxation) is not consistent between different studies and systems, and it may depend on the nature of the sample, aging conditions and/or detection techniques.
Annealing Effect on Free Volume of a Glass
The free volume concept has been used to rationalize stability of a pharmaceutical in a glass, and large free volume is generally associated with high probability of a transport event and therefore high molecular mobility [39]. It is well known that free volume of a glass will decrease during structural relaxation. However, given the challenges in direct experimental measurement of free volume in a glass, particularly in a freeze-dried powder, the correlation of free volume with pharmaceutical stability has not been systematically investigated. Recent gas pycnometry studies on pure trehalose glass showed that density increased significantly (i.e. volume decreased) upon annealing at temperatures higher than 50°C [40], which is consistent with the “densification” effect caused by physical aging [28]. The decrease in volume (and free volume) was also found to correlate with improved chemical stability upon thermal treatment [34, 40], suggesting that free volume measurement may be a simple method for the prediction of long-term stability of pharmaceuticals.
Stabilization of Protein by Annealing
 Previous thermal treatment studies only focused on small molecule pharmaceuticals, and it will be very important to investigate if this annealing can also stabilize protein molecules. One may expect that annealing below Tg will not cause severe protein unfolding in a rigid glass. Previous studies show that the mean structural relaxation time of pure sucrose at 60°C is around 10 hours [29], comparable to the time scale of thermal treatment process. Upon annealing a protein formulation at 60°C for 10 hours, a protein molecule is expected to undergo one molecular “jump”. Therefore, protein unfolding, which is expected to involve a large number of such motions, similar to polymer chain diffusion, would not be expected to proceed significantly. This hypothesis is supported by a recent study which showed that thermal denaturation of hGH in a glassy solid occurs only well above Tg [23].
Previous thermal treatment studies only focused on small molecule pharmaceuticals, and it will be very important to investigate if this annealing can also stabilize protein molecules. One may expect that annealing below Tg will not cause severe protein unfolding in a rigid glass. Previous studies show that the mean structural relaxation time of pure sucrose at 60°C is around 10 hours [29], comparable to the time scale of thermal treatment process. Upon annealing a protein formulation at 60°C for 10 hours, a protein molecule is expected to undergo one molecular “jump”. Therefore, protein unfolding, which is expected to involve a large number of such motions, similar to polymer chain diffusion, would not be expected to proceed significantly. This hypothesis is supported by a recent study which showed that thermal denaturation of hGH in a glassy solid occurs only well above Tg [23].
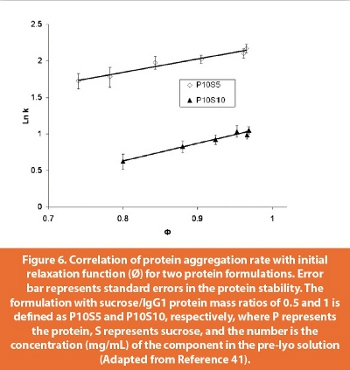
Given the decrease in global molecular mobility upon annealing, protein pharmaceuticals should also be stabilized with a suitable thermal treatment process. The first example of stabilization of protein by annealing has been recently demonstrated using an IgG fusion protein [41]. Two formulations with different IgG/sucrose mass ratios were used, and these samples were subject to different thermal treatment after freeze drying. Annealing increased the storage stability of the protein, as shown by the smaller aggregation rate and lower total aggregation at the end of a storage period. Here, annealing does not significantly impact protein native structure (measured by solid state FTIR) or the local dynamics in the IgG1 protein/sucrose system (studied by both neutron scattering and 13C NMR). Figure 6 shows that the relaxation function measured from DSC correlates quite well with the aggregation rate constant. Therefore, it seems that long-term stability is more sensitive to changes in global mobility in these annealing studies.
Our unpublished data showed similar stabilization effects on rHSA/ sugar formulation upon annealing. The initial aggregate upon thermal treatment at 65°C for 20 hours was still <0.5%, suggesting this approach can be a practical procedure for improving storage stability. More systematic studies are needed to investigate how annealing will impact different protein biopharmaceuticals in amorphous solids. This annealing strategy may have the potential to produce formulations with acceptable long term stability at room temperature storage, and thus eliminate the cold chain requirement of storage condition and enhance the availability of biopharmaceuticals in developing countries.
Summary
Numerous studies have been performed in the field of formulation of lyophilized proteins in the past few decades, and a significant progress has been made to understand the relationship between protein formulation, native structure preservation, and product stability. However, to date, there are insufficient data in the literature to fully define the impact of thermal treatment process on molecular mobility and storage stability. Here, we attempted to develop a useful perspective on the impact of process modulation on global and local mobility and product stability. The temperature variations during freezing, primary and secondary drying were reviewed, and the importance of molecular motions impacting protein stability in the amorphous solid was discussed. Since molecular mobility is significantly impacted by thermal history, variation in lyophilization processing conditions could produce big differences in product stability. It was our aim to provide a picture of the current understanding of the impact of thermal history on product quality so that this knowledge can be used to maximize the pharmaceutical stability using lyophilization process optimization strategy.
Author Biographies
Dr. Bingquan (Stuart) Wang is a Manager in the Commercial Process Development department at Genzyme at Framingham, MA. He received his Ph.D. in pharmaceutics from Michael Pikal’s lab at the University of Connecticut. He has more than 10 years of experience on bioanalytical/ physical characterization of protein, pharmaceutical formulation and fill/finish/Lyo process development. His current responsiblilites include developing scale down models and QbD process design strategies, and providing technical leadership to Technology Transfer to CMO and internal manufacturing sites.
Dr. Pikal is Professor and Distinguished Chair in Pharmaceutical Technology at the University of Connecticut. He received his Ph.D. in physical chemistry in 1966 from Iowa State University. Prior to joining Lilly Research Laboratories in 1972, he was assistant professor of chemistry at the University of Tennessee. Dr. Pikal is a Fellow of the AAPS, and received the AAPS Research Achievement Award in Pharmaceutical Technologies in 2001. His current research activities include the solid state chemistry of pharmaceuticals, particularly the stability of amorphous materials, characterization of solids by calorimetry, and the science and technology of freeze drying with a focus on optimization of formulation and process for labile proteins.
References
1. A.M. Abdul-Fattah, V. Truong-Le, L. Yee, L. Nguyen, D.S. Kalonia, M.T. Cicerone, M.J. Pikal, Drying-induced variations in physico-chemical properties of amorphous pharmaceuticals and their impact on stability (I): Stability of a monoclonal antibody, Journal of Pharmaceutical Sciences, 96 (2007) 1983-2008.
2. A.M. Abdul-Fattah, D.S. Kalonia, M.I. Pikal, The challenge of drying method selection for protein pharmaceuticals: Product quality implications, Journal of Pharmaceutical Sciences, 96 (2007) 1886-1916.
3. S.U. Sane, R. Wong, C.C. Hsu, Raman Spectroscopic Characterization of Drying-Induced Structural Changes in a Therapeutic Antibody: Correlating Structural Changes with Long-Term Stability, Journal of Pharmaceutical Sciences, 93 (2004) 1005-1018.
4. J.F. Carpenter, M.J. Pikal, B.S. Chang, T.W. Randolph, Rational design of stable lyophilized protein formulations: some practical advice, Pharmaceutical Research, 14 (1997) 969-975.
5. M.J. Pikal, Freeze-drying of proteins part II: formulation selection, BioPharm (Duluth, MN, United States), 3 (1990) 26-30.
6. X. Lu, M.J. Pikal, Freeze-drying of mannitol-trehalose-sodium chloride-based formulations: The impact of annealing on dry layer resistance to mass transfer and cake structure, Pharmaceutical Development and Technology, 9 (2004) 85-95.
7. J. Searles, J. Carpenter, T. Randolph, Annealing to optimize the primary drying rate, reduce freezing-induced drying rate heterogeneity, and determine Tg in pharmaceutical lyophilization, J Pharm Sci, 90 (2001) 872-887.
8. M.C. Heller, J.F. Carpenter, T.W. Randolph, Manipulation of lyophilization-induced phase separation: Implications for pharmaceutical proteins, Biotechnology Progress, 13 (1997) 590-596.
9. B. Lueckel, B. Helk, D. Bodmer, H. Leuenberger, Effects of formulation and process variables on the aggregation of freeze-dried interleukin-6 (IL-6) after lyophilization and on storage, Pharmaceutical Development and Technology, 3 (1998) 337-346.
10. J.M. Sarciaux, S. Mansour, M.J. Hageman, S.L. Nail, Effects of buffer composition and processing conditions on aggregation of bovine IgG during freeze-drying, Journal of Pharmaceutical Sciences, 88 (1999) 1354-1361.
11. S.D. Webb, J.L. Cleland, J.F. Carpenter, T.W. Randolph, Effects of annealing lyophilized and spray-lyophilized formulations of recombinant human interferon-γ, J Pharm Sci, 92 (2003) 715-729.
12. X. Tang, S.L. Nail, M.J. Pikal, Freeze-Drying Process Design by Manometric Temperature Measurement: Design of a Smart Freeze-Dryer, Pharmaceutical Research, 22 (2005) 685-700.
13. M. Pikal, J., Freeze drying, in: J. Swarbick, B. Boylan (Eds.) Encyclopedia of Pharmaceutical technology, Marcel Dekker, New York, 2002, pp. 1299-1326.
14. S. Jiang, S.L. Nail, Eff ect of process conditions on recovery of protein activity after freezing and freeze-drying, European Journal of Pharmaceutics and Biopharmaceutics, 45 (1998) 249-257.
15. D.Q. Wang, J.M. Hey, S.L. Nail, Effect of collapse on the stability of freeze-dried recombinant factor VIII and alpha-amylase, Journal of Pharmaceutical Sciences, 93 (2004) 1253-1263.
16. K. Chatterjee, E.Y. Shalaev, R. Suryanarayanan, Partially crystalline systems in lyophilization: II. Withstanding collapse at high primary drying temperatures and impact on protein activity recovery, Journal of Pharmaceutical Sciences, 94 (2005) 809-820.
17. S. Luthra, J.-P. Obert, S. Kalonia Devendra, J. Pikal Michael, Investigation of drying stresses on proteins during lyophilization: differentiation between primary and secondary-drying stresses on lactate dehydrogenase using a humidity controlled mini freeze-dryer, J Pharm Sci FIELD Full Journal Title:Journal of pharmaceutical sciences, 96 (2007) 61-70.
18. J.D. Colandene, L.M. Maldonado, A.T. Creagh, J.S. Vrettos, K.G. Goad, T.M. Spitznagel, Lyophilization cycle development for a high-concentration monoclonal antibody formulation lacking a crystalline bulking agent, Journal of Pharmaceutical Sciences, 96 (2007) 1598-1608.
19. S. Passot, F. Fonseca, N. Barbouche, M. Marin, M. Alarcon-Lorca, D. Rolland, M. Rapaud, Effect of product temperature during primary drying on the long-term stability of lyophilized proteins, Pharm Dev Technol FIELD Full Journal Title:Pharmaceutical development and technology, 12 (2007) 543-553.
20. K. Schersch, O. Betz, P. Garidel, S. Muehlau, S. Bassarab, G. Winter, Systematic Investigation of the Effect of Lyophilizate Collapse on Pharmaceutically Relevant Proteins I: Stability after Freeze-Drying, Journal of Pharmaceutical Sciences, 99 (2010) 2256-2278.
21. M.J. Pikal, S. Shah, M.L. Roy, R. Putman, The secondary drying stage of freeze drying: drying kinetics as a function of temperature and chamber pressure, International Journal of Pharmaceutics, 60 (1990) 203-217.
22. L. Chang, D. Shepherd, J. Sun, D. Ouellette, K.L. Grant, X. Tang, M.J. Pikal, Mechanism of protein stabilization by sugars during freeze-drying and storage: Native structure preservation, specific interaction, and/or immobilization in a glassy matrix?, Journal of Pharmaceutical Sciences, 94 (2005) 1427-1444.
23. M.J. Pikal, D. Rigsbee, M.L. Roy, Solid State Stability of Proteins III: Calorimetric (DSC) and Spectroscopic (FTIR) Characterization of Thermal Denaturation in Freeze Dried Human Growth Hormone (hGH), Journal of Pharmaceutical Sciences, 97 (2008) 5122-5131.
24. M.J. Pikal, K. Dellerman, M.L. Roy, Formulation and stability of freeze-dried proteins: effects of moisture and oxygen on the stability of freeze-dried formulations of human growth hormone, Developments in Biological Standardization, 74 (1992) 21-38.
25. A.M. Abdul-Fattah, R.H. Bogner, M.J. Pikal, The effect of annealing on the stability of amorphous solids: Chemical stability of freeze-dried moxalactam, Journal of Pharmaceutical Sciences, 96 (2007) 1237-1250.
26. S.A. Luthra, I.M. Hodge, M. Utz, M.J. Pikal, Correlation of Annealing with Chemical Stability in Lyophilized Pharmaceutical Glasses, Journal of Pharmaceutical Sciences, 97 (2008) 5240-5251.
27. S.L. Shamblin, G. Zografi , Enthalpy relaxation in binary amorphous mixtures containing sucrose, Pharmaceutical Research, 15 (1998) 1828-1834.
28. I.M. Hodge, Enthalpy relaxation and recovery in amorphous materials, Journal of Non- Crystalline Solids, 169 (1994) 211-266.
29. S.L. Shamblin, X. Tang, L. Chang, B.C. Hancock, M.J. Pikal, Characterization of the Time Scales of Molecular Motion in Pharmaceutically Important Glasses, Journal of Physical Chemistry B, 103 (1999) 4113-4121.
30. M.J. Pikal, Chemistry in solid amorphous matrices: implication for biostabilization, in: H. Levine (Ed.) Amorphous Food and Pharmaceutical Systems, The Royal Society of Chemistry, Cambridge, UK, 2002, pp. 257-277.
31. M.J. Pikal, D. Rigsbee, M.L. Roy, D. Galreath, K.J. Kovach, B.Q. Wang, J.F. Carpenter, M.T. Cicerone, Solid State Chemistry of Proteins: II. The Correlation of Storage Stability of Freeze- Dried Human Growth Hormone (hGH) with Structure and Dynamics in the Glassy Solid, Journal of Pharmaceutical Sciences, 97 (2008) 5106-5121.
32. S.A. Hill, W. MacNaughtan, I.A. Farhat, T.R. Noel, R. Parker, S.G. Ring, M.J. Whitcombe, The Eff ect of Thermal History on the Maillard Reaction in a Glassy Matrix, Journal of Agricultural and Food Chemistry, 53 (2005) 10213-10218.
33. M.J. Pikal, The study of relaxation enthalpy in glassy pharmaceuticals with the thermal activity monitor (TAM). in: Thermometric Seminars on Calorimetry in Materials Sciences., Stockholm, Sweden., 1996.
34. B.Q. Wang, M.J. Pikal, The Impact of Thermal Treatment on the Stability of Freeze Dried Amorphous Pharmaceuticals: I. Dimer Formation in Sodium Ethacrynate, Journal of Pharmaceutical Sciences, 99 (2010) 663-682.
35. S.A. Luthra, M.J. Pikal, M. Utz, Solid state C-13 NMR investigation of impact of annealing in lyophilized glasses, Journal of Pharmaceutical Sciences, 97 (2008) 4336-4346.
36. A.L.M. Smits, F.C. Ruhnau, J.F.G. Vliegenthart, J.J.G. Van Soest, Aging of starch based systems as observed with FT-IR and solid state NMR spectroscopy, Starch/Staerke, 50 (1998) 478-483.
37. J. Perez, J.Y. Cavaille, L. David, New experimental features and revisiting the alpha and beta mechanical relaxation in glasses and glass-forming liquids, J. Mol. Struct. FIELD Full Journal Title:Journal of Molecular Structure, 479 (1999) 183-194.
38. [38] P. Lunkenheimer, R. Wehn, U. Schneider, A. Loidl, Glassy aging dynamics, Physical Review Letters, 95 (2005) 1-4.
39. G. Dlubek, E.M. Hassan, R. Krause-Rehberg, J. Pionteck, Free volume of an epoxy resin and its relation to structural relaxation: Evidence from positron lifetime and pressure-volume-temperature experiments, Physical Review E, 73 (2006).
40. T. Kikuchi, B.. Wang, M.J. Pikal, High-precision absolute (true) density measurements on hygroscopic powders by gas pycnometry: Application to determining effects of formulation and process on free volume of lyophilized products, J Pharm Sci, 100 (2011) pages 2945–2951.
41. B. Wang, M.T. Cicerone, Y. Aso, M.J. Pikal, The Impact of Thermal Treatment on the Stability of Freeze-Dried Amorphous Pharmaceuticals: II. Aggregation in an IgG1 Fusion Protein, Journal of Pharmaceutical Sciences, 99 (2010) 683-700.