Biopharmaceutics, Pharmaceutical Sciences and Clinical Supply
Introduction
Dissolution experiments have been used for years as a quality control tool for pharmaceutical products. The accompanying dissolution specifications are intended to ensure consistency between formulation batches during development and manufacturing. More recently, increased attention has been placed on biorelevant dissolution. Biorelevant dissolution, defined as dissolution in media designed to mimic the composition of the gastrointestinal fluids [1], allows for study of formulation behavior in an environment that could be considered closer to the in vivo conditions compared to compendial media, especially for BCS II/IV drugs. As such, biorelevant dissolution could be seen as more useful in understanding the clinical implications of any formulation changes.
The interplay between API/formulation and oral absorption is complex. Oral absorption of a given compound will be dictated by interactions between a host of factors related both to the gastrointestinal physiology as well as the specific compound/formulation characteristics. Formulation factors such as API phase, API bulk properties (eg. particle size), and presence of surfactants or polymers are commonly explored via dissolution tests to understand their impact on drug solubilization out of the dosage form. In vivo, the impact of these dissolution changes related to formulation may be amplified or attenuated depending on the sensitivity of dissolution process to physiologic parameters such as the different pH in the different GI regions, transit times, administration in the presence of a meal, etc. Given that it would be impractical to fully test these effects in clinical studies for every given formulation of a drug compound, a link needs to be established between the dissolution and the clinical performance.
Traditionally the dissolution – clinical performance link has been sought via the development of an in vitro-in vivo correlation (IVIVC). Establishment of an IVIVC does not necessarily require dissolution in biorelevant media and there is significant experience in its application at least for Modified Release (MR) products. However the development of an IVIVC, especially early in development, is challenging. Significant investment is required as dedicated clinical data on multiple formulations are needed. Development of IVIVC for IR products and especially for BCS II/IV compounds is also extremely challenging; low solubility limits options for dissolution methods and makes the establishment of a correlation between the in vitro and the in vivo data difficult. Given that the majority of products are developed as IR formulations, alternative approaches are needed. In recent years, with the advances in the physiologically-based pharmacokinetic modeling (PBPK), such tools are attracting attention as alternative means to establish this in vitro–in vivo link for formulations.
Oral absorption PbPK modeling
The basic principles of utilization of PBPK modeling to simulate oral absorption have been extensively reviewed in the literature [2-7]. Availability of commercial software has facilitated adoption of these tools during routine pharmaceutical development [3-6]; however many custom models are also utilized and can be customized to provide similar functionalities to commercial solutions [2, 7]. The general concept behind oral absorption modeling is depicted in Figure 1. It involves the simulation of the key processes related to formulation (typically that would be dissolution although more complex processes such as precipitation may need to be accounted for), compound absorption in the gut (permeation process could be passive or active) and drug systemic disposition (with flexibility to utilize compartmental PK or full PBPK models). The simulation takes into account all the relevant GI physiology parameters that could affect drug entry into the body. As an outcome one can get an estimate of either the drug absorption time course or, if of interest, the plasma pharmacokinetics. Given that the simulations are conducted assuming a physiological setting, input of dissolution data under physiological conditions is paramount to the successful use of this approach. Thus biorelevant dissolution represents perhaps the most critical input dictating the accuracy/predictability of the absorption model.
 Figure 1. General Principles of PBPK/Absorption Modeling in Combination with Biorelevant Dissolution Applied to Formulation Development
Figure 1. General Principles of PBPK/Absorption Modeling in Combination with Biorelevant Dissolution Applied to Formulation DevelopmentThe synergy between biorelevant dissolution and PBPK modeling can offer several advantages to formulation/biopharmaceutics scientists. It could provide a mechanistic or at least semi-mechanistic link between dissolution and in vivo response, allowing for rapid assessment of multiple “what if” scenarios to identify key formulation attributes and key experiments. The output of the model may help further refine specific dissolution methods – due to the modular nature of the models, scientists may be able to focus on a simpler dissolution model to study the determining process for absorption. Finally, and perhaps more importantly, it allows for communication of data across scientific principles by translating dissolution and formulation data to a clinical setting. These concepts are demonstrated in the case studies below.
Case study 1: understanding Particle size impact on Dissolution and Pharmacokinetics for a bCs Class ii Compound
The relationship between API particle size and dissolution has been well-documented in the literature and particle size reduction is a commonly employed technique to improve dissolution and exposure of poorly soluble compounds. The use of absorption modeling for understanding impact of API properties has been demonstrated in the literature [8-9].

Compound A is a BCS Class II compound, with moderate solubility of 50 μg/mL in simulated gastric fluid (SGF) and 155 ug/mL in fasted state simulated intestinal fluid (FaSSIF). During development of formulations for FIH studies, jetmilled API was utilized to maximize the dissolution rate and ensure adequate exposures across a wide dose range. The formulation resulted in adequate exposures and linear pharmacokinetics. However, as the compound moved further in development, where doses would cover a narrower range, formulation development needed to account for potential variability in API lots and understand what particle size specifications could be targeted. To guide formulations, absorption PBPK modeling was utilized.
To initiate the modeling, observed dissolution data for jetmilled API and FIH tablets were fitted to a particle-size based dissolution model; subsequently dissolution projections were conducted for a series of test API lots with varying particle size distribution (Figure 2 left panel). At the same time an oral absorption model was built for the FIH data; the model, which was based on the solubility, permeability and API particle size of the compound described the observed data well (Figure 2 right – squares and solid line). With the model at hand, projections were made for the test lots. This allowed for a better understanding of potential variations in exposure by mapping a particle size-exposure space. For example it is demonstrated that if tablets behave like the 33 μm API lot, an approximately 20% loss of Cmax may be seen. With the definition of this space via the PBPK modeling it became evident that larger particle sizes may still be acceptable for the final formulations. Following final experimentation, tablets of particle sizes up to 33 μm continued to result in dissolution within acceptable bounds and were selected for scale up.
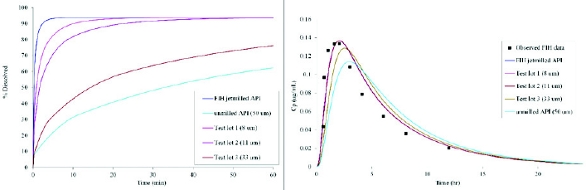 Figure 2. Dissolution modeling (left panel) and associated PK prediction for Compound A. The PBPK model projections allow the definition of an acceptable dissolution (and in this case also particle size) space that can be used to guide further formulation development.
Figure 2. Dissolution modeling (left panel) and associated PK prediction for Compound A. The PBPK model projections allow the definition of an acceptable dissolution (and in this case also particle size) space that can be used to guide further formulation development.While in the case of Compound A, some deviations for Cmax may have been acceptable, the final decision on acceptable operating spaces will be compound-dependent (eg. compounds that require fast onset will be more sensitive to dissolution changes) and also phase of development dependent (if changes had to be made later in development, stricter bioequivalence criteria may have been applied). The PBPK modeling allows to explore these scenarios and guide further experimentation as needed.
Case study 2: guiding Formulation Development for a PPi interaction via PbPK modeling
Compound B is a BCS Class II weak base. It exhibits high solubility in acidic environment (>2 mg/mL at pH 1.2) but very low solubility in FaSSIF (~ 6 ug/mL). Based on the high stomach solubility, a conventional formulation of the free base was utilized for FIH studies, as stomach solubilization was expected to enable sufficient bioavailability. However, an interaction with acid reducing agents was expected. This was confirmed in the early clinical studies. While adequate and linear exposures were observed in the ascending dose study, in subjects dosed with a proton pump inhibitor (PPI), more than 10-fold reduction in exposure was observed [10]. Thus re-formulation efforts were initiated.
To guide formulation development, as previously detailed by Mitra et al [10], first an oral absorption PBPK model was built to describe the observed pharmacokinetics under fasted conditions with and without omeprazole administration (Figure 3 left panel). The model clearly indicated that stomach solubilization was driving exposures. A weak base precipitation as the drug transfers from the stomach to the intestine could be a concern. However in this case, the observed exposure difference suggested that in the naïve fasted state precipitation in vivo is not significant enough to limit exposures. While the input from standard biorelevant media allowed for the description of the observed pharmacokinetics from the FIH formulation, a new “biorelevant” dissolution needed to be developed to simulate the achlorhydric stomach. A low buffer capacity pH 3.0 HCl/NaCl medium was used to screen new formulations – the dissolution in this media was subsequently used for projections of formulation behavior in vivo. The modeling output of select formulations, expressed as the ratio to target Ctrough values, is shown in Figure 3. A few of the formulations were projected to provide adequate bioavailability. The same formulations were screened in parallel in the pentagastrin/famotidine dog model to obtain additional preclinical proof of concept. The outcome of the dog studies was in good agreement with the modeling data; what the modeling data provided in addition was the ability to directly look at the projections against human PK plasma concentration targets. The final formulation that was tested in the clinic showed no significant interaction with a PPI (AUC ratio = 0.95).
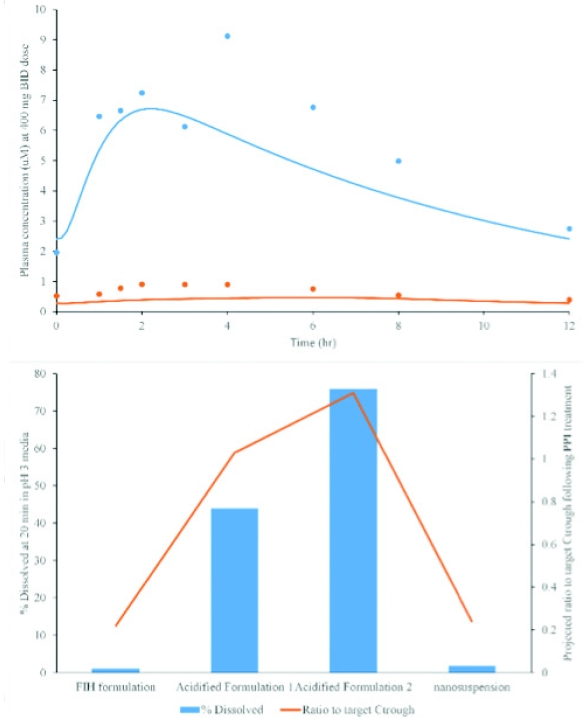 Figure 3. Top: Predicted vs observed clinical exposures from the oral absorption model. Bottom: Predictions of relative exposure (expressed as ratio to Ctrough target) for new test formulations based on dissolution data. Formulation F1 was selected for fi nal development and appeared to mitigate the observed interaction in the clinic. Based on data from Mitra et al [10].
Figure 3. Top: Predicted vs observed clinical exposures from the oral absorption model. Bottom: Predictions of relative exposure (expressed as ratio to Ctrough target) for new test formulations based on dissolution data. Formulation F1 was selected for fi nal development and appeared to mitigate the observed interaction in the clinic. Based on data from Mitra et al [10].Case Study 3: Oral PBPK Modeling for Enabled Formulations – A Case Study for Aprepitant
The increasing percentage of BCS II and IV compounds in drug development pipelines has led to a continuous uptake of new formulation technologies aiming to provide solubilization and absorption enhancement. Liquid-filled capsules, solid dispersions or nanocrystalline suspensions are routinely utilized nowadays during drug development and have led to successful marketed products. Ability to project clinical performance of such formulations is becoming a critical need during product development. However, the dissolution processes for these systems may be more complicated. For example, in lipid systems, lipolysis may affect the in vivo solubilization. In amorphous solid dispersions, several different species, such as nanoparticles or drug-polymer complexes, can be generated, and, in nanosuspensions, the small particle size may further complicate analytical methodologies.
While the field is still evolving, biorelevant dissolution may also have an important role to play in studying these systems. Aprepitant is a BCS Class IV compound marketed as a crystalline nanosuspension formulation in EMEND. Shono et al. [11] have reported how the coupling of biorelevant dissolution data with PBPK modeling could help explain the performance of the nanosuspension formulation relative to the micronized suspension and relative to fasted or fed administration. The authors studied dissolution and solubility of aprepitant in a range of compendial and biorelevant media.
Significantly higher solubilities in biorelevant media were obtained– without accounting for the solubility increase from the bile micelles in vivo, any prediction of pharmacokinetic attempted was poor for either micronized or nanosized aprepitant. Subsequently, the detailed dissolution kinetics were characterized; nanosized API resulted in faster and increased dissolution compared to micronized drug (Figure 4). Incorporating the dissolution rates in a custom-built PBPK model, the authors successfully predicted the relative performance of the formulations with reasonable accuracy.
 Figure 4. Dissolution (left) and fasted state pharmacokinetic projections vs. observed data for micronized and nanosized aprepitant. The combination of biorelevant dissolution data with PBPK modeling successfully mirrored the observed clinical data. Based on data from Shono et al. (11).
Figure 4. Dissolution (left) and fasted state pharmacokinetic projections vs. observed data for micronized and nanosized aprepitant. The combination of biorelevant dissolution data with PBPK modeling successfully mirrored the observed clinical data. Based on data from Shono et al. (11).While successful predictions were demonstrated for aprepitant, it is not clear at this stage if this can be generalized. The exercise was also retrospective with clinical data at hand. The full prediction of plasma concentration profiles for enabled formulation is still generally viewed as challenging especially early in development where clinical data may be limited.
Conclusions and Future Directions
The advances in the biorelevant dissolution field in the recent years have led to new insights to the behavior of compounds and dosage forms in vivo. However the absorption process is quite complex and cannot be easily replicated in a simple dissolution setup. Thus an oral absorption simulation may require development of very complex dissolution systems. PBPK models allow scientists to bring together data from different principles to attempt to put together a more complete picture of the oral absorption for any given compound or formulation. While the knowledge in the field is still growing and new challenges are emerging as formulations also become more complex to deal with the poor solubility of the drug development candidates, there are clear signs on the utility of PBPK models in informing formulation decisions, as demonstrated by relevant publications in the field. Moving forward, ideally development of dissolution methods should take into account the complementary nature of PBPK modeling to allow for design of dissolution systems/assays that would accurately describe disintegration, dissolution and, if applicable, precipitation from a dosage form that would subsequently feed into a PBPK model to allow for simulation of the pharmacokinetic response. While IVIVC establishment will always remain the end goal, the combination of biorelevant dissolution with PBPK modeling may provide alternate means to achieving the IVIVC compared to the traditional methods.
Author Biography
Dr. Filippos Kesisoglou joined Merck in 2005 and is currently leading Modeling & Simulation Quantitative PVE and Oral Biopharmaceutics efforts within the Biopharmaceutics group at West Point, PA. He has supported the biopharmaceutical evaluation of oral formulations across development phases for many PCCs, with special focus on the development and application of absorption modeling and its integration with both preclinical data and with other modeling and simulation tools in a cross-functional setting to facilitate formulation decisions.
References
- Dressman JB, Vertzoni M, Goumas K, Reppas C. Estimating drug solubility in the gastrointestinal tract. Adv Drug Deliv Rev. 2007 Jul 30;59(7):591-602.
- Sugano K. Introduction to computational oral absorption simulation. Expert Opin Drug Metab Toxicol. 2009 Mar;5(3):259-93.
- Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001 Oct 1;50 Suppl 1:S41-67.
- Jamei M, Turner D, Yang J, Neuhoff S, Polak S, Rostami-Hodjegan A, Tucker G. Populationbased mechanistic prediction of oral drug absorption. AAPS J. 2009 Jun;11(2):225-37.
- Thelen K, Coboeken K, Willmann S, Dressman JB, Lippert J. Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part II: extension to describe performance of solid dosage forms. J Pharm Sci. 2012 Mar;101(3):1267-80.
- Johnson KC. Dissolution and absorption modeling: model expansion to simulate the effects of precipitation, water absorption, longitudinally changing ntestinal permeability, and controlled release on drug absorption. Drug Dev Ind Pharm. 2003 Sep;29(8):833-42.
- Sjögren E, Westergren J, Grant I, Hanisch G, Lindfors L, Lennernäs H,Abrahamsson B, Tannergren C. In silico predictions of gastrointestinal drug absorption in pharmaceutical product development: Application of the mechanistic absorption model GI-Sim. Eur J Pharm Sci. 2013 Jul 16;49(4):679-98.
- Kesisoglou F, Wu Y. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 2008 Dec;10(4):516-25.
- Zhang X, Lionberger RA, Davit BM, Yu LX. Utility of physiologically based absorption modeling in implementing Quality by Design in drug development. AAPS J. 2011 Mar;13(1):59-71.
- Mitra A, Kesisoglou F, Beauchamp M, Zhu W, Chiti F, Wu Y. Using absorption simulation and gastric pH modulated dog model for formulation development to overcome achlorhydria effect. Mol Pharm. 2011 Dec 5;8(6):2216-23.
- 11. Shono Y, Jantratid E, Kesisoglou F, Reppas C, Dressman JB. Forecasting in vivo oral absorption and food effect of micronized and nanosized aprepitant formulations in humans. Eur J Pharm Biopharm. 2010 Sep;76(1):95-104.