Introduction
Orally disintegrating tablets (ODT) or sublingual technologies have drawn attention in the industry as a promising approach to deliver drugs in life cycle management to a wide range of the patient population [1-6]. Those technologies are also aimed at patients incapable of swallowing pills, especially in pediatric and geriatric populations [7]. The criteria for rapid dissolution and onset of active ingredients in an ODT dosage depends on the performances of individual excipients which must qualify 3 unique criteria: (i) hydrophilic in nature, (ii) dissolves quickly with good mouth feel, and (iii) is highly compressible. Identifying such excipients and/or technologies to meet these criteria could be challenging, especially to overcome the drug’s loading and stability.
Currently, a wide spectrum of innovative, directly compressible co-processed excipients is available to meet these criteria for an ODT [8]. The manufacturers of these excipients continue to invest in technologies to meet the challenges stemming from solubility and/or delivery of APIs in nearly all areas of therapeutic developments. These therapeutic areas can include analgesics (non-steroidal and opioids), antimigraine, antianginal, antiemetics, tranquillizers, antipsychotics, antihistaminics, antispasmodics, antitussives, glucocorticoids, hypnotics/ sedatives, and vasodilators among others. Likewise, contract research and manufacturing organizations (CROs/CMOs) have developed their own proprietary technologies to meet the challenges in this rapidly growing area. Those technologies give formulators the opportunity to design and develop the ODTs more cost-effectively [3].
The examination of the products in Table 1 warrants further evaluation to better understand the excipients and processes for ODT. This review is aimed at understanding the ODT compositions and the roles of excipients play in design and disintegration of such dosage forms.
Table 1. Marketed OdT Apis*

Composition of an ODT
In an ODT composition, the hydrophilic and lipophilic characteristics of excipients with abilities to dissolve, rapidly absorb water and swell to yield pressure for disintegration are highly desired. This article will focus on the key ingredients responsible for disintegration and quick onset resulting from water absorption, and will also shed light on understanding the underlying mechanisms responsible for drug release and stability of ODT dosages.
Process of Manufacturing an ODT Dosage
Figure 1 illustrates the formation of an ODT co-processed composition typical derived from mannitol (Table 1), and a disintegrant. The process requires the granulation of mannitol and disintegrant with an appropriate binder. The resulting granules are highly porous which, on exposure to water, rapidly disintegrate the tablet, allowing the API to disperse and dissolve in the saliva of mouth.
 Figure 1. Illustration of an ODT dosage preparation and after disintegration/dissolution.
Figure 1. Illustration of an ODT dosage preparation and after disintegration/dissolution.This article will highlight a co-processed excipient (referred to as Mannitol/PVAc/PVP) comprised of mannitol, NF, 90%; crospovidone, NF, 5%; and polyvinyl acetate 30% dispersion, NF, and polyvinylpyrrolidone, NF together at 5% [9]. The polyvinyl acetate dispersion is used as a granulating agent that provides binding of the granules by direct compression. These attributes are critical in development of an ODT formulation.
Brief Methodology
The performance, stability, flowability, use of disintegrants, effect of lubricants, disintegration methods, and effect of packaging of the coprocessed Mannitol/PVAc/PVP excipient were evaluated in placebo and model drugs.
Results and discussions
Mannitol/PVAc/PVP ODT Placebo Dosages
Figure 2 illustrates the tablet hardness, friability and disintegration time as a function of compression force, kN. The placebo tablet (10 mm in diameter) contained 98% Mannitol/PVAc/PVP and 2% sodium stearyl fumarate, and weighed 300 mg each, and compressed at 4.8 kN with hardness of 44 N yielding friability <0.2%. The Mannitol/PVAc/PVP excipient hardness and disintegration time increased as the compression forces increased, while the friability decreased. The ODT tablets showed a disintegration time of 19 seconds and 15 seconds in healthy volunteers.
 Figure 2. Compression profile of Mannitol/PVAc/PVP placebo ODT.
Figure 2. Compression profile of Mannitol/PVAc/PVP placebo ODT.Mannitol/PVAc/PVP ODT with Model Drugs
Several model drugs have been evaluated with the Mannitol/PVAc/PVP in formulations by direct compression, but this review will be restricted to one model drug.
Table 2 shows the composition of cetirizine dihydrochloride formulation. In the formulation preparation, API was granulated with Mannitol/PVAc/ PVP and saccharine with 6.5% PVP K25 solution in a fluid bed granulator with atomizing pressure of 0.5 bar, inlet air temperature of 45°C to 50°C and outlet temperature of 30°C. The resulting granules were post-granulated with additional Mannitol/PVAc/PVP and blended with peppermint aroma flavor and magnesium stearate as lubricant, and sieved through 0.8-mm screen before compression at 2 kN.
Table 2. Composition and properties of Cetirizine OdT

Stability of ODT Dosage Forms
The stability of all the ODT dosage forms can be challenging depending upon drugs and excipients used in the formulations. It is recommended to store the dosages at temperatures ≤25°C and use a dense packaging material to control the humidity and moisture permeation. Hence, the Mannitol/PVAc/ PVP ODTs were packaged in PVC/PVDC blisters. Briefly, the tablets were compressed with hardness of 54 N, and each tablet weighed 300 mg, and had a diameter of 10 mm with flat surface. The placebo tablets contained 98% Mannitol/PVAc/PVP and 2% sodium stearyl fumarate as a lubricant.
Mannitol/PVAc/PVP placebo ODTs were subjected to stability test which was carried out at 30°C (drying oven) and at 23°C at 33% to 90% relative humidity (RH). The stability test results are shown in Figure 3. Disintegration time (DT) remained almost unchanged when the tablets were exposed to relatively higher humidity conditions at 23°C. On exposure to higher temperature, the DT increased significantly. Consequently, the hardness of tablets also increased at 30°C but remained unchanged over a 3-month period at 23°C at different %RH (data not shown here). These results reveal that simple placebo tablets are more sensitive to temperature than to humidity. However, the impact of temperature on DT can be mitigated by the addition of disintegrants to the tablet formulation. This can be noted in the loperamide ODT formulation which consisted of 2.0 mg loperamide, 134.5 mg Mannitol/PVAc/PVP, 15.0 mg disintegrant, 2.0 mg chocolate flavor and 3.0 mg sodium stearyl fumarate compressed into 8-mm tablets of approximately 40N hardness at a compression force of 5kN. The tablets had been stored in closed polyethylene bottles for 12 months under relevant ICH climate conditions. Figure 4 illustrates that DT remained constant independent of the storage conditions.
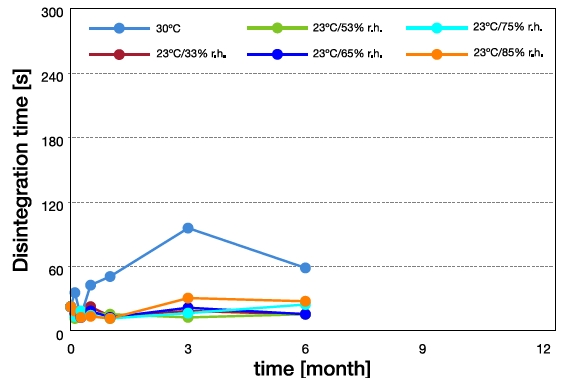 Figure 3. Disintegration of placebo ODTs as a function to time at 30°C (drying oven) and 23°C (%RH’s) conditions.
Figure 3. Disintegration of placebo ODTs as a function to time at 30°C (drying oven) and 23°C (%RH’s) conditions. Figure 4. Disintegration of loperamide ODTs as a function to time at different climate conditions.
Figure 4. Disintegration of loperamide ODTs as a function to time at different climate conditions.Flowability of Mannitol/PVAc/PVP ODT Formulation
Compression aids usually possess an excellent flowability and, when used with other excipients and drugs, may cause segregation during tableting. This is of particular relevance because drugs used in ODTs are typically lowdosed and thus difficult to homogeneously blend. The Mannitol/PVAc/PVP excipient has a medium flowability as can be recognized from the angle of repose (38°C to 39°C). However, if flowability needs to be further improved, colloidal silica can be added. Table 3 shows the data with and without colloidal silica on the performance of the placebo ODT which contained 2% sodium stearyl fumarate as a lubricant.
Table 3. Effects of colloidal silica on OdT dosages
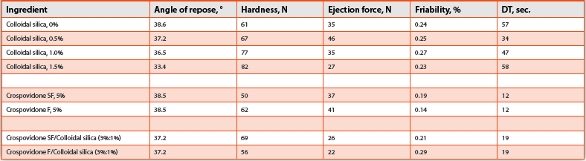
With all additives tested the hardness remained at a high level and the ejection force at a very low level indicating that all formulations show a good potential for tablet forming and no sign of stickiness.
Effects of Disintegrants on ODT Dosages
The disintegrants strongly determine the performances of the ODT dosages. The porosity of an ODT formulation plays an important role which allows the permeation of water molecules for the required disintegration. Koizumi et al. [10] increased the porosity by using camphor as a sublimating agent in the mannitol-based ODT. However, disintegrants are commonly used in many of the ODT formulations. Therefore, a careful identification and selection of a disintegrant is critical for the development and long-term stability of the tablets. The physico-chemical properties of each disintegrant depend upon the chemistry and inherent porosity of the polymeric molecules [11]. For instance, Mishra et al. [12] studied a range of disintegrants on the disintegration of valdecoxib and metoclopramide model drugs in ODT formulations including croscarmellose sodium, and sodium starch glycolate (SSG), and found that the DT was dependent upon the disintegrants used. Porosity and water absorption rates affect disintegration. Faster disintegration is associated with rapid water absorption, and increased pressure due to capillary action of micropores in water insoluble polymeric matrix.
The functionalities of disintegrants are also dependent on the drugs and other ingredients in an ODT. In an acetylsalicylic acid formulation containing 5% acid-treated yeast as a binder with different disintegrants, carmellose considerably reduced the disintegration time (ca. 20 seconds) as compared to croscarmellose sodium, carmellose calcium and low subsitituted hydroxylpropyl cellulose (L-HPC) [13]. Such differences were due to faster swelling and less gelling characteristics of carmellose than other disintegrants. In an earlier study, Watanabe et al. [14] observed that L-HPC in meclizine ODT with MCC as a binder, significantly reduced the disintegration time (ca. ≤30 seconds), especially with 10% to 30% finer grade of the disintegrant (<50 microns). In a recent study, Venkatesh et al. [15] examined the effects of finer grade crospovidone in a pulsatile multiparticulate system comprised of mannitol, and observed that the tablets were robust when hydroxpropyl cellulose grade applied as coating around the beads. The data with Mannitol/PVAc/PVP, however, reveal that the additional amounts of finer grade disintegrants can shorten the disintegration time even when tablets are exposed to higher temperatures and humidities as shown in Table 3 and Figures 3 and 4.
Roblegg et al. [16] studied Mannitol/PVAc/PVP as a binder in wet granulation of paracetamol and ibuprofen, and found that the drug release profiles were dependent upon the solvents and pH conditions. The data from this study also suggest that Mannitol/PVAc/PVP mixed with cellulosic excipients as binders can enhance the mechanical stability of pellets and provide a good mouth feel and faster release profile. The faster disintegration was also noted in melt granulation of acetaminophen with glyceryl palmitostearate as a binder, and sodium starch glycolate as a disintegrant. The disintegration time was significantly reduced with as low as 5% of ATO binder, and the drug release attained over 80% in 10 minutes [17].
Although the ODT formulation dosages are aimed primarily at delivery of water soluble drugs, several insoluble compounds have been developed. For instance, carvedilol, a poorly soluble crystalline drug, has been investigated in mannitol-based ODT formulations to yield the faster disintegration by converting into an amorphous solid dispersion [18]. In contrast, when benzocaine, an insoluble drug, was examined in Mannitol/ PVAc/PVP ODT formulations, the appearance, performance, and stability of tablets were of higher quality as compared to other mannitol-based excipients [19]. Ironically, the performances of ODT dosages could be related to a drug’s specificity and compatibility in the formulations. Kruppa et al. [20] observed that the formulations containing ibuprofen, diclofenac sodium and dilitiazem were critical on the performance of ODT dosages, and were also dependent upon solubility and loading of drugs. In our studies, when loratadine, a poorly soluble (aq. solubility <1 μg/ml) antihistamine drug was used with Mannitol/PVAc/PVP, the in vitro and in vivo disintegration times were 38 and 32 seconds, respectively (unpublished). However, the use of disintegrants can further reduce the disintegration time to 5 to 10 seconds.
Effects of Lubricants on ODT Dosages
The effects of lubricants on an ODT are not fully understood. Only a few commonly used lubricants have been studied including sodium stearyl fumarate, magnesium stearate, stearic acid, and poloxamers. The recommended levels of these lubricants are usually between 0.5% and 2%. In a recent study, Kruse et al. [21] investigated the effects of 2 different lubricants on Mannitol/PVAc/PVP ODT, and found that the wetting property and lipophilicity of sodium stearyl fumarate play a role in reducing the disintegration. Thus, the disintegration time was 17 seconds with sodium stearyl fumarate as compared to 28 seconds with 2% magnesium stearate, and the tablets were relatively harder under similar conditions. In a recent study, Kuno et al. [22, 23] investigated talc as a lubricant in an ODT compared with commonly used lubricants such as sodium stearyl fumarate and magnesium stearate. The authors reported that talc performed better and significantly reduced the disintegration times of sugar alcohols’ ODTs due to increased hydrophilicity onto the outer surface of matrix, thus allowing greater water permeation into the tablets. In our studies when sodium stearyl fumarate and magnesium stearate were used in Mannitol/PVAc/PVP ODT formulations, sodium stearyl fumarate used as low as 1% significantly increased the ejection force to 142 N versus 51 N at 2% of the same and compromised the tablet hardness and the stickiness. Disintegration time and friability were unaffected (data not shown). Our study demonstrates that the use of a lubricant with at least 2% Mannitol/PVAc/PVP composition will significantly reduce ejection force.
Disintegration Method for ODT Dosages
The FDA recommends using the method as outlined for ODT dosages in the USP Chapter <701>. In a typical procedure, the ODTs are subjected to in vitro dissolution tests in the USP Type I (basket) and/or the USP Type 2 dissolution apparatus with and without disk and sinkers at low to medium paddle speed of about 50 rpm [24]. It is often challenging to find the appropriate dissolution test method for an ODT and is still debatable [25]. However, the USP II remains the preferred method over Type 1 [26]. There are, however, alternative approaches to alleviate the differences between the conventional USP and in vivo test methods. For instance, Park et al. [27] proposed a simulated wetting test (SWT) method as an alternative to the USP ODT method. The SWT method showed a stronger correlation with in vivo disintegration of acetaminophen ODT in healthy volunteers as compared to conventional USP II method. There is certainly an advantage in selecting one method over another depending upon the formulations, and selection is even more critical when developing a dietary supplement ODT [28]. Therefore, the in vivo mouth testing of an ODT is highly recommended for development of an ODT dosage. The in vivo tests could be subjective depending upon the geographical population and age group. To alleviate such variations, a number of instrumental devices are also available. For example, a device equipped with 8-mm diameter flat-faced punches is available for testing ODTs. This device is sensitive to upper punch displacement, relaxation time of upper punch displacement, and relaxation time of die wall force during compaction [29]. Harada et al. [30, 31] developed an ODT in vitro test method for disintegration of an ODT dosage to mimic the human sensory test. In our studies with Mannitol/ PVAc/PVP placebo samples, the disintegration times were approximately the same (15 to19 seconds) when tested in vitro or in vivo.
Packaging of ODT Dosages
A majority of the ODT dosages usually suffer from longer disintegration time due to instability of formulation resulting in an increased hardness on storage in an uncontrolled and/or under ambient and/or accelerated conditions [6]. This leads to a slower release and is often associated with excipients’ properties and/or an API's incompatibility in the formulations. Moisture absorption could also lead to a slower release but, if controlled by using the appropriate packaging material, the stability of API can be improved. The Mannitol/PVAc/PVP excipient is no different than others in the market, and requires stringent moisture controls. Therefore, appropriate packaging is required to increase the shelf life of drug products. Alu-Alu, Aclar-Alu and PVC-PVDC-Alu packaging materials are highly recommended for blistered packaging to protect from moisture uptake. Desiccants are also effective for the same with high density polyethylene containers.
Conclusions
This article presents a modern perspective on ODT technologies, and highlights the roles of excipients in formulation design and development to address the challenges of stability. Our mannitol-based co-processed Mannitol/PVAc/PVP excipient can support water soluble and insoluble compounds for the development of ODTs. The Mannitol/PVAc/PVP formulation data indicate that the performance and stability of an ODT dosage can be adjusted or modified to meet the criteria for disintegration time by additional amounts of finer grade crospovidones. The process aids like colloidal silica, sodium stearyl fumarate, magnesium stearate, and talc among others can be used as glidants and lubricants to enhance the performance of the dosages and their processibility. Appropriate packaging is equally critical for long term stability and performance of the ODT dosages.
References
- D. Brown, Orally disintegrating tablets-taste over speed. Drug Del. Technol. 2003, 3, 58–61.
- Y. Fu, S. Yang, S. H. Jeong, S. Kimura, and K. Park, Orally fast disintegrating tablets: Developments, technologies, taste-masking and clinical studies, Critical. Rev. Therap. Drug Carrier Systems, 2004, 21, 433-475.
- R. McLaughlin, S. Banbury, and K. Crowley, Orally Disintegrating Tablets, The Effect of Recent FDA Guidance on ODT Technologies and Applications, Pharm. Tech., September 2009.
- J. J. Hirani, D. A. Rathod, and K. R. Vadalia, Orally disintegrating tablet: A review, Tropical J. Pham. Res., 2009, 8, 161-172.
- E. Kumar and J. Bhagyashree, Mouth dissolving tablets, Int. J. Pharm. Res. Rev., 2013, 2, 25-41.
- S. Velmurugan and S. Vinushitha, Orally disintegrating tablets: An overview, Int. J. Chem. Pharm. Sci., 2010, 1, 1-12.
- T. Shu, H. Suzuki, K. Hironaka, and K. Ito, Studies of rapidly disintegrating tablets in the oral cavity using co-ground mixtures of mannitol with crospovidone, Chem. Pharm. Bull., 2002, 50, 193-198.
- K. Shahani, N. Deorkar, and A. Gandhi, Tablets & Capsules, Eye on Excipients, March 2011.
- BASF: Ludiflash®-Technical Brochure, March 2012.
- K. Koizumi, Y. Watanabe, K. Morita, N. Naoki, and M. Matsumoto , New method of preparing high-porosity rapidly saliva soluble compressed tablets using mannitol with camphor, a subliming material, Int. J. Pharm., 2009, 152, 127-131.
- R. Pahwa and N. Gupta, Superdisintegrants in the development of orally disintegrating tablets: A review, Int. J. Pham. Sci Res., 2011, 2, 2767-2780.
- D. N., Mishra, M. Bindal, S. K. Singh, and S. Gurusam, V. Kumar, Spray Dried Excipient Base: A novel technique for the formulation of orally disintegrating tablets, Chem. Pharm. Bull, 2006, 54, 99-102.
- T. Ozeki, Y. Yasuzawa, H. Katsuyama, Y. Takashima, T. Kasai, T. Eguchi, H. Kakiuchi, H. Yuasa, and H. Okada, Design of rapidly disintegrating oral tablets using acid treated yeast cell wall: A technical note, AAPS PharmSciTech, 2003, 4, 1-4.
- Y. Watanabe, K. Koizumi, Y. Zama, M. Kiriyama, Y. Matumoto, and M. Matsumoto, New compressed tablet rapidly disintegrating in saliva in the mouth using crystalline cellulose and a disintegrant, Biol. Pharm. Bull., 1995, 18, 1308-1310.
- G. M. Venkatesh, P. J. Stevens and J-w. Lai, Development of orally dinitegrating tablets comprising controlled release multiparticulate beads, Drug. Dev. Indust. Pharm., 2012, 38, 1428-1440.
- E. Roblegg, S. Schrank, M. Griesbacher, S. Radl, A. Zimmer, and J. Khinast, Use of the direct compression aid Ludiflash® for the preparation of pellets via wet extrusion/spheronization, Drug Dev. Indust. Pharm, 2011, 1-13.
- G. A. Popa, L. Ochiuz, I. Stoleriu, I. Cojocaru, and I. Popovici, Formulation and preparation of orally disintegrating tablet using innovative binder, Farmacia, 2013, 61, 1131-1136.
- R. N. Shamma and M. Basha, Soluplus®: A novel polymeric solubilizer for optimization of carvedilol solid dispersions: Formulation design and effect of method of preparation, Powder Tech., 2013, 237-406-414.
- M. Kollmer, C. Popesen, P. Manda, L. Zhou, and R. A. Gemeinhart, Stability of benzocaine formulated in commercial oral disintegrating tablet platforms, AAPS PharmSciTech, 2013, 14, 1333-1340.
- Krupa, R. Jachowicz, Z. Pedzich, and K. Wodnicka, The influence of the API properties on the ODTs manufacturing from co-processed excipient systems, AAPS PharmSciTech, 2012, 13, 1120-1129.
- S. Kruse, S. Gebert, K. Meyer-Böhm, A. Maschke and K. Kolter, Compression Characterization and Lubricant Sensitivity of Orally Disintegrating Tablets Based on Ludiflash® , APV World Meeting 2008, 7.-10. April 2008, Barcelona, Spain.
- Y. Kuno , M. Kojima, S. Ando, H. Nakagami, Effect of preparation method on properties of orally disintegrating tablets made by phase transition, Int. J. Pharm., 2008, 355, 87-92.
- Y. Kuno, M. Kojima, H. Nakagami, E. Yonemochi, and K.Terada, Effect of the type of lubricant on the characteristics of orally disintegrating tablets manufactured using the phase transition of sugar alcohol, Eur. J. Pharm. Pharmacol., 2008, 69, 986-992.
- J. Klancke, Dissolution testing of orally disintegrating tablets, Dissolution Technologies, May 2003, 6-8.
- K. Schmid and R. Lobenberg, Influence of the changed USP specifications on disintegration test performance, Dissolution Technologies, February 2010, 6-10.
- G. Dromgoole, Distek, personal communication.
- J. H. Park, K. M, Holman, G. A. Bish, D. G. Krieger, D. S. Ramlose, C. J. Herman, and S. H. Wu, An alternative to the USP disintegration test for orally disintegrating tablets, Pharm. Tech., 2008, 54-58.
- M. Almukainzi, M. Salehi, N. A. Bou-Chacra, and R. Lobenberg, Investigation of the performance of the disintegration test dietary supplements, The AAPS J., 2010, 12, 602-607.
- Y. Shibata, Y. Yamamoto, M. Fuji, M. Kondoh, and Y. Watanabe, A novel method for predicting disintegration time in the mouth of rapidly disintegrating Tablet by compaction analysis using TabAll, Chem. Pharm. Bull., 2004, 52, 1394-1395.
- T. Harada, R. Narazaki, T. Ohwaki, T. Uchida, Effect of physical properties of orally disintegrating tablets on disintegration time as determined by a new apparatus, J. Drug Del. Sci. Tech., 2010, 20, 377-383.
- T. Harada, R. Narazaki, S. Nagira, T. Ohwaki, S. Aoki, and K. Iwamoto, Evaluation of the disintegration properties of commercial famotidine 20 mg orally disintegrating tablets using a single new test and human sensory test, Chem. Pharm. Bull., 2006, 54, 1072-1075.
Shaukat Ali has over 20 years of experience in the pharmaceutical industries including 10 years at BASF, where he supports the solubilization platform. His areas of expertise include modified release, film development, liposome drug delivery, and solubilization technologies. He serves on the editorial advisory board of American Pharmaceutical Review and other publications including Contract Pharma, Drug Development & Delivery, Biopharma Asia (UK), and International Journal of Pharmaceutical Investigation. He is also a member of the USP panel of experts for General Chapters-Physical Analysis. He received his Ph.D. in Chemistry from the City University of New York and carried out his postdoctoral work at the University of Minnesota and Cornell University. He has authored over 25 scientific articles and is inventor in 14 US patents. He is an adjunct faculty at the College of Pharmacy of University of South Florida.
Karl Kolter obtained his Ph.D. from the Johannes Gutenberg University of Mainz and worked thereafter for 7 years at a pharmaceutical company where he was involved in the development of oral liquid drugs, parenterals, sustained release drug delivery systems and drugs produced by direct compression. In 1993, he joined BASF SE, where he was responsible for R&D in pharmaceutical excipients, drug formulations and the application technology of vitamins and carotenoids for pharmaceuticals and food. The focus of his current work is the development of innovative excipients, mainly for solid oral dosage forms. This work has already resulted in a number of new products. In 2010, he was honored with the Award for Industry Research in Excipient Technology and in the same year one of his latest developments received the CPhI Silver Innovation Award. Karl Kolter has published more than 140 articles and posters and is inventor of more than 90 patents.