Antibody-Drug Conjugates – the next generation of chemotherapy
The ability to discriminate and eliminate cancerous cells without harming healthy tissues has long been a goal in cancer therapy. Antibody-drug conjugates (ADCs) advance this goal by exploiting the targeted specificity of an antibody to deliver a highly-potent therapeutic agent; thus, bypassing the unwanted side effects associated with conventional chemotherapies.1* An ADC is composed of three fundamental elements: the monoclonal antibody, which selectively binds cancer-specific antigens; the cytotoxic drug payload, which induces cell death; and the small molecule linker, which connects the previous two units (Figure 1A). As demonstrated in Figure 1B, ADCs bind to their cognate antigen, are internalized by endocytosis, and traffic to lysosomes where the cytotoxic payload is released, e.g., via proteolytic cleavage. Then, the released payload diffuses into the cytoplasm or the nucleus to bind its designated target and trigger cell death. The coupling of the protein and small molecule elements promises to increase the potency of antibody therapeutics and mitigate the toxicity associated with high-potency small molecules. Thus, ADCs represent a new enhanced biologic with an expanded therapeutic window, offering increased potency with decreased toxic side-effects.
 Figure 1. Key parameters of antibody-drug conjugates A) Components of site-specifically modified antibody-drug conjugate. B) Mechanism of drug delivery. C) Drug to antibody ratios and hypothetical drug distribution of lysine, cysteine and site-specifically conjugated ADCs.
Figure 1. Key parameters of antibody-drug conjugates A) Components of site-specifically modified antibody-drug conjugate. B) Mechanism of drug delivery. C) Drug to antibody ratios and hypothetical drug distribution of lysine, cysteine and site-specifically conjugated ADCs.Current ADCs and their limitations
Currently, there are two FDA-approved ADCs, Adcetris™ and Kadcyla™. At the molecular level, both of these therapeutics are heterogeneous mixtures resulting from the nonselective ligation of drug to native cysteine and lysine residues, respectively.2* The nonselective ligation approaches used to make these two ADCs render it very difficult to optimize their biological, physical, and pharmacological properties as any surface-accessible residue can serve as a site of conjugation. This lack of selectivity affords products with various drug loadings, yielding drug-to-antibody ratios (DARs) of 0-8 (Figure 1C). In addition to variable drug loading, conjugation occurs at multiple residues, yielding ADC constructs that are a mixture of isoforms. For example, peptide mapping has shown that approximately 20 different lysines (40 per antibody) are conjugated, resulting in more than one million possible isoform permutations.3*. This immense complexity makes analytical characterization a challenge. These mixtures are usually studied as an ensemble, therefore all efficacy, pharmacokinetic (PK), and toxicity data represent a mean value of the population. The approval of Adcetris™ and Kadcyla™ is a boon for both the patients treated by these therapeutics and the field of ADCs. However, there is still significant room for improvement in controlling heterogeneity, payload loading, and lot-to-lot reproducibility during manufacturing.
Site-Specific ligation chemistries to overcome limitations of conventional heterogeneous conjugations.
To combat these difficulties groups have begun employing site-specific ligation chemistries. Site-specificity creates near homogeneous conjugates in a reproducible fashion by conjugating a known number of drugs to a defined location on the antibody. This method allows detailed study of efficacy and pharmacokinetics (PK) based on DAR and drug placement location without the need for complex analytics. Moreover, there is growing evidence that site-specific conjugation improves PK and safety profiles.4* As a result, there is an incredible investment in the creation of site-specific technologies and a growing body of literature highlighting these new techniques.5* There are chemistries, such as PLP, that modify the N-terminal amino acid allowing for selective conjugation at the N-terminus.6* In addition, there are chemistries that incorporate selenocysteines at the C-terminus and therefore modify that site.7* Derivatization of the antibody’s pendant glycans can be performed by several methods. For example, sialic acid can be enzymatically installed and then treated with periodate to yield an aldehyde, which can react with aminooxy compounds to form oximes.8* Additionally, there are several methods for incorporating an azido sugar into the glycan; then, the azido sugar can be conjugated using copper-free click chemistries. An enzymatic method takes advantage of transglutaminase to conjugate a native glutamine (Q295) to aliphatic amines bearing azides, which are then ligated to payload via copper-free click chemistry. Unfortunately, this conjugation technique can only be performed after removal of the antibody’s glycans.9* All of these chemistries occur at immutable positions on the antibody. They are site-specific, but not positionally programmable.
Currently, there are four major techniques that allow programmable payload placement: engineered cysteine (Genentech/Seattle Genetics), unnatural amino acids (Ambrx, Sutro), chemoenzymatic conjugation (Rinat-Pfizer), and SMARTag™ technology (Catalent), an aldehyde tag-based conjugation. The engineered cysteine technique uses cysteine-maleimide chemistry and requires careful selection of the new cysteine’s location in order to prevent disulfide shuffling or protein dimerization.10,4,11* Unnatural amino acid incorporation uses new modified amino acid substrates, which contain a bioorthogonal functional group that can be incorporated into antibodies using an evolved tRNA/aminoacyl-tRNA synthetase pair. The amino acids are a p-acetylphenylalanine and p-azidomethylphenylalanine, which are conjugated via an oxime ligation 12* or by copper-free click chemistry,13* respectively. Strop and co-workers from Rinat-Pfizer developed a five amino acid tag, LLQGA that can be incorporated into a variety of sites and allows for the direct attachment of payloads to the glutamine via a bacterial transglutaminase.14*
SMARTag™ technology also incorporates an enzymatic step to site-specifically install a chemical handle for subsequent bioconjugation.15* Specifically, the formylglycine generating enzyme, which is endogenous to mammalian cells, is used to site-specifically generate a bioorthogonal functional group, an aldehyde, during protein production.16* This aldehyde is then ligated using aldehyde-specific chemistries (Figure 2).17*
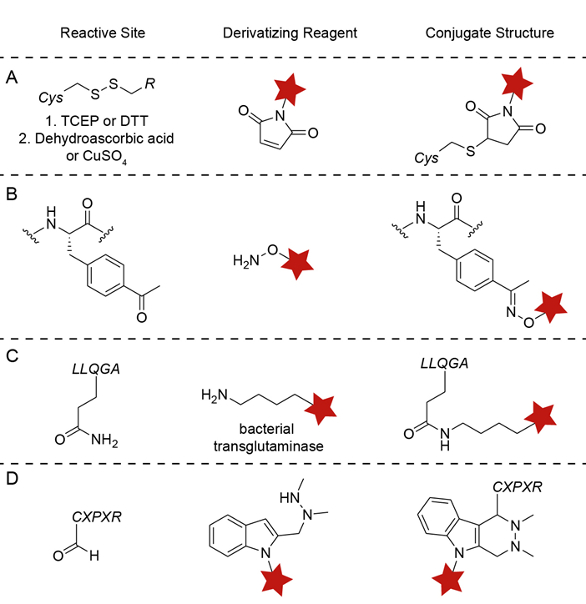 Figure 2. Chemistries for site-specific protein modification. A) Engineered cysteine. B) Unnatural amino acid – p-acetylphenylalanine. C) Bacterial transglutaminase. D) SMARTagTM – HIPS and aldehyde tag substrates.
Figure 2. Chemistries for site-specific protein modification. A) Engineered cysteine. B) Unnatural amino acid – p-acetylphenylalanine. C) Bacterial transglutaminase. D) SMARTagTM – HIPS and aldehyde tag substrates.SMARTag™ technology generates site-specific ADCs
Redwood Bioscience, recently acquired by Catalent Pharma Solutions, pioneered a unique chemoenzymatic method using the naturally occurring endogenous formylglycine-generating enzyme (FGE). FGE cotranslationally converts the cysteine in the pentapeptide sequence CXPXR, where X represents any neutral amino acid residue, to a formylglycine (fGly) residue.18* During protein production FGE oxidizes the cysteine in the consensus sequence to formylglycine. This co-translational modification removes the need to generate and purify a second recombinant enzyme in addition to the protein of interest. The cysteine in the aldehyde tag pentapeptide is converted with exquisite fidelity and allows the aldehyde tag to be placed at a variety of sites on the protein and retain high conversion to fGly (>95%)17* with exceptional protein titers (5g/L) across a variety of tag placements. This combination of positional programmability and prodigious protein production makes the SMARTag™ unique in the ADC space (Figure 3).
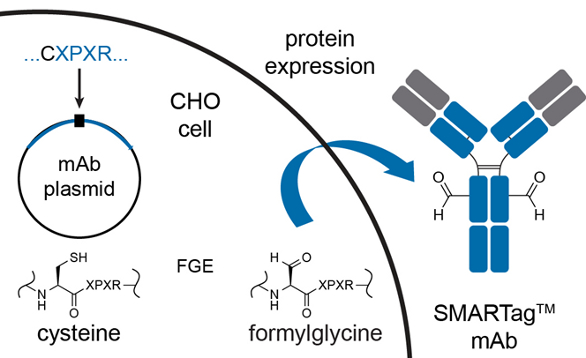 Figure 3. Creation of aldehyde-tagged antibody using formylglycine generating enzyme.
Figure 3. Creation of aldehyde-tagged antibody using formylglycine generating enzyme.Aldehyde-tagged ADCs using iso-Pictet-Spengler chemistries
Historically, oximes and hydrazones have been used for ligation to carbonyls. These alpha-effect nucleophiles possess many of the desired characteristics for bioconjugation; namely, ease of synthesis and selective reactivity with carbonyls in aqueous media. However, oximes have two major drawbacks. First, oximes and hydrazones have a labile C=N bond that is subject to hydrolytic instability. It has been shown in our lab19,20* and others21* that fGly-oximes are hydrolytically unstable and possess a serum half-life of less than 18 h. While this maybe adequate for short-term laboratory studies, the poor serum stability renders this ligation technique a liability for ADCs. This instability could lead to premature payload loss and result in unintended toxicity. Second, oxime formation requires acidic conditions (pH 4.5) to proceed at any appreciable rate and even under ideal conditions the reaction often takes days.12* Prolonged exposure to acid may damage sensitive biomolecules and may exclude some potential payloads from use.22-24*
In order to overcome the instability of oxime constructs, we sought a means to form a more stable bond. We hypothesized that a C-C bond would provide the requisite serum stability for use in ADCs. The classic Pictet-Spengler reaction forms a C-C bond between tryptamine and an aldehyde; however, it employs strong acids like hydrochloric acid in conjunction with elevated temperatures.25* These classical conditions are simply not amenable to protein bioconjugation. Seeking to revamp the reaction and make it more biologically compatible, we utilized an alpha-effect nucleophile to mitigate the harsh acidic conditions and moved the substitution of the indole to the 2-position in order to free the highly nucleophilic 3-position of the indole. These modifications allow the reaction to be performed in water; this reaction has been dubbed the iso-Pictet-Spengler (PS) ligation (Figure 4A). The rate can further be improved by 30% with the addition of an electron-donating methoxyl group to the 5-position of the indole.21* Fluorescence polarization analysis of an AlexaFluor488-containing PS construct conjugated to an aldehyde-tagged maltose binding protein (fGly-MBP) demonstrated no deconjugation in PBS over 7 days. While the new C-C bond formed in the PS ligation is more than adequate for use with ADCs, the PS ligation still suffers from the need to be run under acidic conditions.
 Figure 4. HIPS chemistries generate stable C-C bonds site-specifically. A) HIPS and aza-HIPS proceed through the same hydrazonium intermediate to form a stable C-C bond (red). B) Overview of the HIPS ligation to an aldehyde-tagged antibody. C) ELISA-based assays testing the stability of ADCs conjugated via oxime and HIPS in human plasma at 37°C. D) HPLC analysis can be used to monitor a conjugation reaction proceeding from an unconjugated, aldehyde-tagged antibody (DAR = 0, left) to a conjugated ADC (DAR ~ 2, right).
Figure 4. HIPS chemistries generate stable C-C bonds site-specifically. A) HIPS and aza-HIPS proceed through the same hydrazonium intermediate to form a stable C-C bond (red). B) Overview of the HIPS ligation to an aldehyde-tagged antibody. C) ELISA-based assays testing the stability of ADCs conjugated via oxime and HIPS in human plasma at 37°C. D) HPLC analysis can be used to monitor a conjugation reaction proceeding from an unconjugated, aldehyde-tagged antibody (DAR = 0, left) to a conjugated ADC (DAR ~ 2, right).To further increase the utility of the PS ligation, we sought to optimize the rate of reaction at near neutral conditions. It has been observed that optimal rates of addition to carbonyls occur when the pH is close to the pKa of the protonated nucleophile.26* By replacing the aminooxy component of the PS construct (pKa ~ 4.8 for alkoxyammonium ions) with an alkyl hydrazinyl nucleophile (pKa ~ 6.6 for trialkylhydrazonium ions), the bioconjugation, now a hydrazinyl iso-Pictet-Spengler (HIPS), can be run at neutral or near neutral conditions (pH 6.0-7.0), opening the door to more pH-sensitive payloads and proteins.27*,28* We found the HIPS construct to be a very versatile ligating agent across a multitude of proteins and aldehyde-tag positions. 19,29*(Figures 4A and 4B)
In this vein, a variety of different payloads, including microtubule disruptors and DNA alkylators, have been successfully conjugated using HIPS chemistry. Attachment of HIPS to a payload is done via peptide coupling, where an amine-bearing payload is coupled to an Fmoc-protected HIPS-acid, synthesized in six simple steps from commercially available material. Once the payload is coupled, deprotection of the Fmoc group affords the ligation-ready HIPS-payload construct.
ADCs generated using the HIPS construct show excellent stability in human plasma (Figure 4C). By contrast to the oxime, which had only 40% of the payload remaining after 18 h at 37 °C, the HIPS construct showed no loss of payload by 18 h. Furthermore, when the experiment is extended to two weeks, only minimal loss of conjugate is observed. Additionally, we have developed a more water-soluble version of the HIPS construct, aza-HIPS, that carries a nitrogen substitution at the 7 position in the benzene ring (Figure 4A). The aza-HIPS construct exhibits plasma stability similar to the HIPS construct.30* Use of aza-HIPS is recommended for hydrophobic payloads. The programmability of the aldehyde tag when used in conjunction with the highly aldehyde-specific HIPS chemistries creates homogeneous SMARTag™ ADCs. These ADCs, as shown in Figure 4D, have well controlled DARs and exhibit no payload/linker cross-reactivity with other functionalities on the protein. These data were further corroborated by high-resolution mass spectrometry.
ADCs generated using the SMARTag™ technology have demonstrated high potency (sub-nanomolar range) in vitro across a variety of targets and cell lines, and have demonstrated efficacy against in vivo tumor xenografts. In head-to-head in vivo mouse model experiments, we observed that αHER2 SMARTag ™ ADCs with a DAR of 1.7 were equally efficacious as compared to a conventional lysine conjugate, αHER2-DM1, with double the drug loading (DAR of 3.5).29* In rat PK studies, SMARTag ™ ADCs outperformed ADCs generated by either conventional lysine or cysteine conjugation (Figure 5A). The total antibody-drug conjugate half-lives of the conventional lysine and cysteine conjugates were significantly shorter as compared to the SMARTag ™ ADCs, supporting previous observations that the nonspecific conjugation chemistries (which lead to overconjugated species) have a negative effect on PK.31* Furthermore, toxicological studies were performed in rats to compare the tolerability of a SMARTag™ ADC to a heterogeneous lysine MCC-DM1 conjugate (Figure 5B). Following published protocols,32* we performed a single dose study with an intravenous dose given at 0, 6, 20, and 60 mg/kg. As previously observed, all of the animals died when given a 60 mg/kg dose of the DM1 construct. By contrast, the SMARTag™ ADC was tolerated at all doses, with only reversible changes observed in the hematopoietic and clinical chemistry panels.
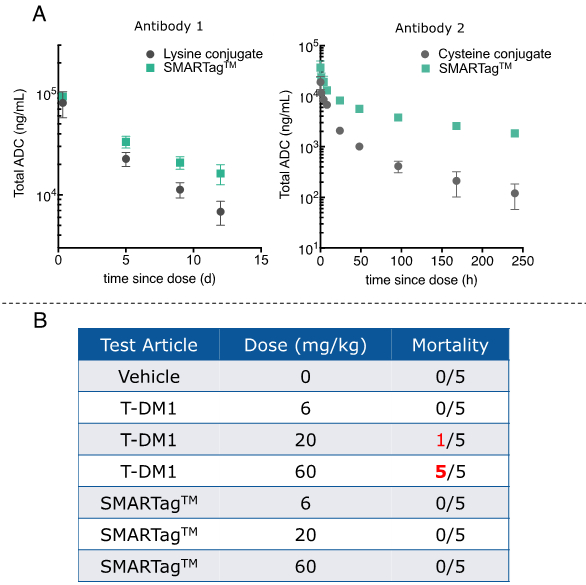 Figure 5. Pharmacokinetic and safety data indicate that SMARTagTM ADCs outperform ADCs generated via conventional conjugation chemistries. A) Rat PK data showing that SMARTag TM ADCs have longer ADC half-lives than either a lysine conjugate (left panel) or a cysteine conjugate (right panel). B) SMARTag TM ADCs were tolerated in rats at all doses tested, by contrast to the lysine MCC-DM1 conjugate, which was lethal at 60 mg/kg.
Figure 5. Pharmacokinetic and safety data indicate that SMARTagTM ADCs outperform ADCs generated via conventional conjugation chemistries. A) Rat PK data showing that SMARTag TM ADCs have longer ADC half-lives than either a lysine conjugate (left panel) or a cysteine conjugate (right panel). B) SMARTag TM ADCs were tolerated in rats at all doses tested, by contrast to the lysine MCC-DM1 conjugate, which was lethal at 60 mg/kg.In summary, the aldehyde tag used in conjunction with HIPS-based chemistries generates highly homogeneous ADCs. The mildness of the HIPS chemistry allows it to be used with a variety of different payloads and proteins to generate stable, biophysically well-behaved ADCs that offer excellent in vivo efficacy, PK, and safety profiles.
References
- Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. doi:10.1146/annurev-med-050311-201823.
- Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. mabs. 2013;6(1):34–45. doi:10.4161/mabs.27022.
- Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23(9):1137–1146. doi:10.1038/nbt1141.
- Junutula JR, Raab H, Clark S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26(8):925–932. doi:10.1038/nbt.1480.
- Agarwal P, Bertozzi CR. Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjugate Chem. 2015:150130084838000. doi:10.1021/bc5004982.
- Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS, Francis MB. N-terminal protein modification through a biomimetic transamination reaction. Angew Chem Int Ed Engl. 2006;45(32):5307–5311. doi:10.1002/anie.200600368.
- Hofer T, Skeffington LR, Chapman CM, Rader C. Molecularly defined antibody conjugation through a selenocysteine interface. Biochemistry. 2009;48(50):12047–12057. doi:10.1021/bi901744t.
- Zhou Q, Stefano JE, Manning C, et al. Site-Specific Antibody–Drug Conjugation through Glycoengineering. Bioconjugate Chem. 2014;25(3):510–520. doi:10.1021/bc400505q.
- Gauthier L, Lhospice F, Romagné F. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody–drug conjugates. Bioconjugate …. 2014.
- Junutula JR, Bhakta S, Raab H, et al. Rapid identification of reactive cysteine residues for site-specific labeling of antibody-Fabs. J Immunol Methods. 2008;332(1-2):41–52. doi:10.1016/j.jim.2007.12.011.
- Jeffrey SC, Burke PJ, Lyon RP, et al. A Potent Anti-CD70 Antibody–Drug Conjugate Combining a Dimeric Pyrrolobenzodiazepine Drug with Site-Specific Conjugation Technology. Bioconjugate Chem. 2013;24(7):1256–1263. doi:10.1021/bc400217g.
- Axup JY, Bajjuri KM, Ritland M, et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc Natl Acad Sci U S A. 2012;109(40):16101–16106. doi:10.1073/pnas.1211023109.
- Zimmerman ES, Heibeck TH, Gill A, et al. Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconjugate Chem. 2014;25(2):351–361. doi:10.1021/bc400490z.
- Strop P, Liu S-H, Dorywalska M, et al. Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chemistry & Biology. 2013;20(2):161–167. doi:10.1016/j.chembiol.2013.01.010.
- Rabuka D. Chemoenzymatic methods for site-specific protein modification. Current Opinion in Chemical Biology. 2010;14(6):790–796. doi:10.1016/j.cbpa.2010.09.020.
- Rush JS, Bertozzi CR. New Aldehyde Tag Sequences Identified by Screening Formylglycine Generating Enzymes in Vitroand in Vivo. J Am Chem Soc. 2008;130(37):12240–12241. doi:10.1021/ja804530w.
- Rabuka D, Rush JS, deHart GW, Wu P, Bertozzi CR. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat Protoc. 2012;7(6):1052–1067. doi:10.1038/nprot.2012.045.
- Carrico IS, Carlson BL, Bertozzi CR. Introducing genetically encoded aldehydes into proteins. Nat ChemBiol. 2007;3(6):321–322. doi:10.1038/nchembio878.
- Agarwal P, Kudirka R, Albers AE, et al. Hydrazino-Pictet-Spengler Ligation as a Biocompatible Method for the Generation of Stable Protein Conjugates. Bioconjugate Chem. 2013:130603084658007. doi:10.1021/bc400042a.
- Kudirka R, Barfield RM, McFarland J, et al. Generating Site-Specifically Modified Proteins via a Versatile and Stable Nucleophilic Carbon Ligation. Chemistry & Biology. 2015:1–7. doi:10.1016/j.chembiol.2014.11.019.
- Agarwal P, van der Weijden J, Sletten EM, Rabuka D, Bertozzi CR. A Pictet-Spengler ligation for protein chemical modification. Proceedings of the National Academy of Sciences. 2012.
- Kee J-M, Muir TW. Chasing phosphohistidine, an elusive sibling in the phosphoamino acid family. ACS Chem Biol. 2012;7(1):44–51. doi:10.1021/cb200445w.
- Devlin GL, Chow MKM, Howlett GJ, Bottomley SP. Acid Denaturation of alpha1-antitrypsin: characterization of a novel mechanism of serpin polymerization. J Mol Biol. 2002;324(4):859–870. doi:10.1016/S0022-2836(02)01088-4.
- Curry S, Abrams CC, Fry E, et al. Viral RNA modulates the acid sensitivity of foot-and-mouth disease virus capsids. J Virol. 1995;69(1):430–438.
- Tatsui G. Uber die Synthese von Carbolinderivaten.
.Yakugaku Zasshi. 1928;48:453–459. - Jencks WP. Studies on the mechanism of oxime and semicarbazone formation1. J Am Chem Soc. 1959.
- Hinman R. Notes-base strengths of some alkylhydrazines. J Org Chem. 1958.
- Bissot TC, Parry RW, Campbell DH. The physical and chemical properties of the methylhydroxylamines1. … American Chemical Society. 1957.
- Drake PM, Albers AE, Baker J, et al. Aldehyde Tag Coupled with HIPS Chemistry Enables the Production of ADCs Conjugated Site-Specifically to Different Antibody Regions with Distinct in Vivo Efficacy and PK Outcomes. Bioconjugate Chem. 2014;25(7):1331–1341. doi:10.1021/bc500189z.
- Albers AE, Garofalo AW, Drake PM, et al. European Journal of Medicinal Chemistry. European Journal of Medicinal Chemistry. 2014:1–7. doi:10.1016/j.ejmech.2014.08.062.
- Hamblett KJ, Senter PD, Chace DF, et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clinical Cancer Research. 2004;10:7063–7070.
- Poon KA, Flagella K, Beyer J, et al. Preclinical safety profile of trastuzumab emtansine (T-DM1): mechanism of action of its cytotoxic component retained with improved tolerability. Toxicol Appl Pharmacol. 2013;273(2):298–313. doi:10.1016/j.taap.2013.09.003.

Dr. Romas Kudirka graduated from University of California, San Diego with a BS in Chemistry. As one of the first chemists at Genomics Institute of the Novartis Research Foundation, he worked on the development of privileged nucleoside libraries. Returning to complete his PhD in Organic Chemistry at the University of California, Irvine under the tutelage of Prof. David Van Vranken, his research involved the discovery of new reactions utilizing palladium-carbene intermediates generated from diazo compounds. Dr. Kudirka’s post-doctoral work focused on the mechanism of self-assembly of biologically inspired polymers. He currently works as a chemist at Catalent Pharma Solutions, formerly Redwood Bioscience, as a Senior Research Scientist, where he specializes in the development of new aldehyde conjugation techniques; linker strategy and design; and synthesis of highly potent cytotoxic payloads.