Abstract
Numerous elements are needed to bring a promising therapeutic protein from research through development, analysis, manufacturing, and quality control (QC) and assurance (QA) for a successful IND application. These include recombinant expression of the protein pharmaceutical in a production cell line, optimization of production, development of a rigorous purification process to achieve ppm levels of impurities, analytical assay development and qualification, scale-up to cGMP manufacturing from lab-scale, testing and documentation to support quality control and assurance, and aseptic fill/finish to create the final drug product.
Introduction
Filing an Investigational New Drug (IND) application with the FDA or other regulatory bodies is one of the most critical steps in developing a new biopharmaceutical. For many start-ups, it is a make-or-break milestone that if not reached can result in substantial loss of support and even closure. Regulatory acceptance of the IND is what allows a sponsoring company to test its new biopharmaceutical in patients for a First-in-Human (FIH) clinical trial. Without clinical data from these studies showing safety and efficacy of a new biopharmaceutical, it has minimal value. Accordingly, the process of molding an initial discovery of a promising new protein into an actual biopharmaceutical for clinical evaluation must be well planned and thorough. This review will outline key steps in the process, which will be described in the Chemistry, Manufacturing and Controls Section (CMC) of the IND. It is intended as a guideline rather than an all-inclusive, detailed review.
Complexity of Proteins
 Figure 1. Etanercept (Enbrel®) Fusion Protein showing multi-chain structure, disulfide bridges and glycosylation sites (public domain image).
Figure 1. Etanercept (Enbrel®) Fusion Protein showing multi-chain structure, disulfide bridges and glycosylation sites (public domain image).Proteins are inherently much more complex than chemical drugs for a host of reasons. First, they are large polymers made by linking together chains of amino acids, and are typically 1000x larger than a chemical drug. The protein chains are made up of one of twenty (20) natural amino acids at each position. The selection and order of these amino acids determines the primary structure of the protein. These chains adopt localized structures in space, such as helices or beta sheets, which are termed the secondary structure and are dependent on the primary structure. As these chains lengthen the protein adopts a three-dimensional structure, called the tertiary structure, typically arranging in the most energetically favorable state. Finally, many proteins contain more than one chain, covalently bound or non-covalently associated, into a larger multi-chain structure, the quaternary structure.
Secondly, proteins often have additional structures covalently attached to them known as post-translational modifications (PTMs).
The most common PTM is carbohydrate, with the resulting protein becoming a glycoprotein. Like proteins these types of carbohydrates are also complex, made from chains of sugar molecules arranged both linearly and in a branched fashion, and with linkage variability as alpha or beta configurations. Other types of PTMs include hydroxylation of lysine or proline side chains, carboxylation of glutamic acid side chains to add an additional, gamma carboxyl group, and phosphorylation of tyrosine residues. All of these PTMs add significant complexity to an already complex protein structure. Figure 1 shows a complex, four-chain protein with extensive glycosylation as the PTMs.
Recombinant Production of Protein
Classically protein pharmaceuticals were isolated directly from natural sources, such as animal organs or human blood. This approach was often fraught with challenges of crude and variable starting materials, low amounts of the desired protein, and safety in terms of microbiological contaminants. Today modern protein pharmaceuticals are produced instead by recombinant techniques, which means one or more gene(s) coding for the protein of interest is inserted into the chromosomes of a production cell. This technology has evolved so substantially that it is now possible to produce high titers of the desired protein from cells grown in chemically defined, serum-free media.
One of the first decisions to make is the choice of expression cell line to produce the recombinant protein. First, this depends on the types of PTMs, if any, that are present on the protein. The simplest production cells, bacteria such as E. coli, cannot make any PTMs. Thus, for a simpler protein such as insulin, which lacks PTMs, bacteria may be good choice for host cell. Their advantages include very rapid growth, relatively simple media, and robustness. However, recombinant proteins made in bacteria are often found in inclusion bodies within the bacterial cell, from which they must be extracted and refolded into the correct conformation. There are some bacterial systems that preferentially express the protein into the periplasmic space (Pseodomonas fluorescens1), or actually secrete it (Corynabacterium glutamacium2), aiding in the recovery of the desired protein. However, gram-negative bacteria such as Escherichia or Pseudomonas also produce endotoxin, which must be rigorously removed from the protein of interest.
Next up the evolutionary ladder is yeast, which is widely used for production of some recombinant proteins in strains designed for this purpose. Yeast also grow rapidly, secrete the protein of interest and are robust organisms. They will also glycoyslate, but produce a primitive, high-mannose form of N-linked glycosylation. Through a series of elegant gene knockout and insertion studies, the company Glycofi was able to engineer their yeast strain to produce full mammalian, human-like glycosylation3. However, this technology has not been widely adopted, in spite of being acquired by Merck.
For production of glycoproteins, and other proteins with PTMs, the cell lines of choice have been mammalian. While much slower growing and more delicate than bacteria and yeast, mammalian cells make a full range of PTMs, and conveniently secreted the recombinant protein fully folded into the culture media. One particular cell line, Chinese Hamster Ovary (CHO) cells, has dominated the production of therapeutic glycoproteins4. CHO cells are well-characterized, reasonably robust for a mammalian cell, have been adapted to grow in suspension, and have available many chemically defined media and feeds that allow high levels of production. Indeed, thanks to advances in the understanding of the growth needs and genetics of mammalian cells, especially CHO cells, production titers have climbed more than 100X on the past twenty years.
There are certainly alternative mammalian cells, including other rodent cells like murine NS0 and hybridomas, in which a number of protein therapeutics are produced. There are also fully human cells such as PER.C6 and HEK cells. These can be desirable in applications where purely human glycosylation5, as opposed to that produced by other mammalian cell lines, is important to the properties of the protein.
Finally, there are alternative cell-lines that have specialized applications.
Production in insect cells, for example, is conveniently done using a baculovirus to carry in the gene for the protein of interest while the virus infects the cell. This technology is used widely in research, since small quantities of proteins can be made quickly. It is also convenient for vaccines that need to be reformulated each year, such as the influenza vaccine, since the new genes can readily incorporated into the baculovirus6.
Regardless of the choice of cell line, production of a recombinant protein begins with transfection of the host cell line with genetic material for the protein of interest. The resulting mixed population of cells is separated into individual clones through any of several possible techniques, such as limit-dilution, cell sorting, or soft-agar cloning.
Cells are selected on the basis of their growth rates, productivity, ability to grow to high densities, and genetic stability over many (>50) generations. The amount of expressed recombinant protein produced in a bioreactor, which is called the titer, is a function of the specific productivity, i.e., how much each cell is producing per unit time interval, multiplied by the cell densities over the time of the bioreactor run. Higher titers translate into lower cost of goods for the product, since smaller and few bioreactor runs are needed to produce the same amount of material.
Typically one selects a lead clone and a couple of back-up candidates for “banking”, which involves aliquoting the cloned cells into several hundred small cryovials. Specialized cryopreservation techniques and media are used to preserve cell viability during the freezing process, storage at liquid-nitrogen temperatures and subsequent thaw as needed for production. Initially smaller research banks are produced for use in development. Cell banks for use in manufacturing human pharmaceuticals, Master Cell Banks (MCBs) and Manufacturer’s Working Cell Banks (MWCBs), must be rigorously safety tested according to regulatory guidelines7.
 Figure 2. Photo comparing shake-flask to small-scale bioreactor cultures.
Figure 2. Photo comparing shake-flask to small-scale bioreactor cultures.Following selection of the cell line, growth conditions for the cell line are optimized to maximize productivity. Multiple parameters are assessed, from selection of the basal culture media to feed solutions used to replenish nutrients consumed by the growing cells. Initial studies are typically carried out in shake-flask cultures, followed by small-scale bioreactors. At the bioreactor stage one can also more precisely control pH, CO2, oxygen, temperature, and agitation rate (see Figure 2). More recently there has been a trend towards the use of automated arrays of mini-bioreactors with volumes as low as 10 mL to allow more in-depth evaluation of such bioreactor conditions8,9.
There has been enormous progress in the past 25 years in developing chemically define media and feed solutions to support vigorous and sustained growth of cells for production of proteins.
Finally, following growth of the production cells in a bioreactor or fermentor, the recombinant protein must be recovered from the complex mixture of cells, cell debris and media. For cells that secrete the protein of interest, this is a matter of removal of the cells and cell debris using filtration and/or centrifugation. For production systems where the desired protein is still within the cells or cell organelles, the solids are isolated from the culture media and processed further as a paste from which to extract the protein. In either case the resulting filtrate, supernatant or extract serves as the feedstock for the purification process.
Protein Purification
Besides the protein of interest, production cells also produce their own proteins, which are termed host-cell proteins (HCP), along with DNA, RNA, and multiple other substances needed by living cells. Thus, the desired protein, whether retained by the cell or secreted, will be accompanied by multiple other components that must be removed via protein purification techniques. These are largely based on the twin techniques of filtration and chromatography. Filtration includes depth, micro, ultra and nanofiltration techniques that separate by size, either to pull out the protein or the impurities. Chromatography, depending on the type, exploits multiple properties of the desired protein, including specific binding, charge, hydrophobicity, charge distribution, and size.
In developing a suitable purification method, one applies the principles of separation science under conditions that preserve the activity and properties of the protein10. Highly selective affinity interactions between the protein and a modified support, such as between antibodies and Protein A resins, provide especially powerful separation of the desired protein from other components in the mixture. The distribution and types of charges on a protein, as well as its isoelectric point, form the basis for the widely used technique of ion-exchange chromatography. Proteins also vary considerably in hydrophobicity, allowing separation by hydrophobic-interaction chromatography. Newer chromatographic modalities rely on combinations of these techniques in one, resulting in what has been termed “mixed-mode” chromatography. The classic technique of size exclusion chromatography, while not as scalable as other methods, still finds use in some applications.
The developer of a protein purification process has the challenge of achieving highly purified material from a rather crude feedstock, the bioreactor harvest. Since the product is almost certain to be an injectable pharmaceutical due to the nature of a protein, the standards for purity are stringent, allowing only ppm levels of impurities.
Furthermore the process must be suitable for larger scale production in a reasonable timeframe in manufacturing11. The application of newer, high-speed filters and chromatographic supports, as well as higher capacity ones, has been valuable in this regard. More recently the introduction of new functionalized membranes, which incorporate charge or other modifications, have presented high-speed membrane separations as alternatives to chromatography. Regardless of the process step, conditions must be selected that avoid degradation and aggregation of proteins, some of which unfortunately show tendencies toward these adverse effects.
Analytical Evaluation and Testing
Throughout the development of the cellular production and protein purification process, one must constantly assess the quantity, quality and purity of the recombinant protein. From the outset this begins as simply method(s) to determine the amount or titer of the protein produced by different cells and growth conditions. These are followed by testing to determine the quality of the protein in terms of its structure and function. As noted earlier, proteins are sensitive macromolecules and as such are subject to multiple routes of thermal and enzymatic degradation, as well as denaturation and aggregation.
Bioanalytical assays serve as the “eyes and ears” of the process development scientist developing a process for the production and purification of the desired protein. They answer key questions such as how much protein is being produced, what quality it is, and how pure it is at each stage of the purification process.
A series of analytical techniques are used to test and characterize a protein pharmaceutical. These assays can be categorized as to what characteristic is being tested. First there are assays for molecular and structural integrity. Aggregation is most commonly measured by size exclusion chromatography (HPLC-SEC), which can also measure to a limited extent peptide-bond cleavage. A stronger technique for this is denaturing electrophoresis, typically run as gels (SDS-PAGE) or capillary electrophoresis (CE-SDS). Detection of charge variants, such as those from deamidation, requires charge-sensitive techniques such as ion exchange chromatography (IEX) and isoelectric focusing in gels (IEF) or capillaries (CE). Protein mass and post-translational modifications typically require mass spectrometry and peptide-mapping HPLC.
The second category of assays measures the amount of protein, both in terms of concentration and activity or potency of the protein drug. Because most proteins contain aromatic amino acids, ultraviolet spectroscopy (UV) is nearly always used to quantitate purified proteins. In crude states, such as bioreactor harvest, the amount of protein produced is measured either by chromatography or Enzyme-Linked Immuno-Sorbet Assays (ELISA). The actual activity or potency of a protein is measured according to its key properties.
Binding molecules, such as monoclonal antibodies, must be capable of binding their target structures consistently and with high affinity.
Enzymes are measured for their enzymatic activity, usually with a colorimetric assay utilizing a chromogenic substrate. Often such assays have an ELISA format in microtiter plates, allowing multiple samples and replicates for accuracy and precision. Some protein pharmaceuticals may be inhibitors or stimulators of binding by other molecules, such as growth factors. This type of activity may require cell-based assays, in which the effect of the test protein on the growth and properties of the cell is assessed.
The final category of biopharmaceutical assays measures the presence of impurities in the protein preparation. These include host cell proteins, as well as nucleic acids and other components from the host cell. Additionally, some types of purification techniques, such as Protein A chromatography and other protein-based affinity techniques, can shed their affinity ligands into the process. These types of process residuals must also be measured, typically using a ligand-specific ELISA. Finally, there are also product-related impurities, such as degradants from the protein of interest.
Analytical techniques are also needed to assess the stability of the purified protein over time. Proteins degrade at rates depending on their intrinsic qualities, storage temperature, presence of impurities, the solution in which they are formulated, and the container in which they are stored. Indeed, the development of formulations to minimize degradation, to maintain vital qualities of the protein, is a vital part of the development process. It is complex and really a separate science in itself, utilizing sophisticated techniques such as Differential Scanning Calorimetry (DSC), chromatographic and electrophoretic techniques to assess the stability of the protein in different formulations.
A complex glycoprotein such as an IgG antibody presents multiple sites for instability. These are illustrated in Figure 3. Some of these sites, such as truncation of the labile C-terminal lysine residues of the heavy chains, generally do not change the activity or efficacy of this protein drug. Other changes may result in substantial effects on the molecule, such as amino-acid residue changes in or near the antigen-binding site of the antibody. Aggregation is a special concern, since it is well known that a protein’s immunogenicity increases when aggregated.
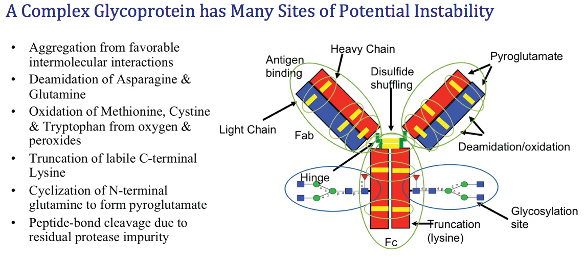 Figure 3. Potential sources of instability on an IgG antibody. Graphic courtesy of Dr. Greg Kilby, Agilent Technologies, used with permission.
Figure 3. Potential sources of instability on an IgG antibody. Graphic courtesy of Dr. Greg Kilby, Agilent Technologies, used with permission.These molecular changes to a protein over time must be assessed for their effects on its safety and efficacy as a biopharmaceutical; hence, the need for strong analytics supporting stability studies.
Scale-Up and Manufacturing Under cGMP
Once a process for production, purification and analytical testing is established, the process must be scaled up to manufacturing.
A well-developed process is one that will scale successfully, with consideration given to the larger volumes and sizes of manufacturing.
There are both scientific/technical and quality/regulatory aspects to the scale-up process. Scientifically the process must be one that replicates at a scale typically at least 100X larger than the developed bench-scale process. Standard principles of chemical engineering are applied to ensure that vital parameters like fluid mixing and gas uptake in the bioreactor, and filter capacities, are similar across scales.
Downstream chromatography columns are scaled by residence time to ensure reproducible separation as columns get larger. Sufficient tankage must be available for the increasing volumes of media, buffers and product fractions.
On the quality/regulatory side, the new process must be one that meets the requirements of current Good Manufacturing Practices (cGMP), as defined by regulatory authorities. While a discussion of cGMP is beyond the scope of this article, one vital element is that all processes must follow pre-written and approved procedures, such as Standard Operating Procedures (SOPs) and Master Batch Records (MBRs). Critical equipment should be validated for its intended use, and cleaning procedures for reuse of equipment as well. In this later regard, the widespread application of single-use systems throughout biomanufacturing has lessened the burden of cleaning studies, as there are no risks for cross-contamination if the fluid-contact surfaces of a piece of equipment are single-use.
A pilot or engineering run is invaluable in scaling from say a 10L bench scale process to a 1000-2000L cGMP production process. It serves as a critical run-through of the process at larger scale, both for the process as well as the produced and purified protein product. Furthermore it is a valuable opportunity to hone and refine a process one final time before the cGMP runs. Draft batch records can be used and edited and improved, and operators can be trained on the process. In this regard it is valuable to have the process-development scientists who developed the process work side-by-side with their manufacturing colleagues. In spite of best efforts to clearly explain a process in writing, there is no substitute for hands-on training. Both development and manufacturing scientists benefit. The development scientists see first-hand what is involved at larger scale, while the manufacturing scientists learn the critical aspects of any particular process.
Quality Control (QC) and Quality Assurance (QA)
These critical elements of pharmaceutical manufacturing involve testing (QC) and documentation (QA) for all processes. QC testing extends from raw materials and environmental monitoring through in-process and final bulk drug substance and drug product testing.
All test methods or assays must be run according to documented and approved test procedures, which are at least qualified, if not validated for the samples being tested. Quality Assurance governs all the documents of a cGMP process, from raw materials through to final product, including controlled access, review and final approval.
Special Safety Studies for Biopharmaceutical
Besides the testing of the cell banks (MCB & MWCB) mentioned earlier, additional safety testing is required of the cells following a manufacturing run. On the downstream side, viral-clearance studies are required of any mammalian-cell process to prove that the purification process can remove potential viruses, either endogenous ones such as retroviruses or adventitious ones that may be inadvertently acquired during a process. Such viral-clearance studies are based on appropriate scaled-down processes in which small quantities of test viruses are added to representative feedstocks for those steps in the downstream process expected to either inactivate or remove viruses.
Guidelines for such studies are available from both the FDA12 and European13 regulatory authorities.
Drug Product
Protein therapeutics are nearly always administered by injection, since they would not survive the harsh conditions of the gastrointestinal track. Therefore, the final drug product is a parenteral pharmaceutical.
Because proteins also cannot tolerate terminal sterilization processes such as autoclaving, the final drug product is aseptically filled under stringent conditions to minimize potential microbial contamination.
For early-phase studies, the most common format is a liquid formulation in sterile injection vial, typically stored either refrigerated (2-8°C) or frozen (-20 or -70°C), depending on the product stability. For some sensitive proteins, it may be necessary to develop a freeze-dried or lyophilized formulation that would be reconstituted as a liquid just prior to patient administration. More recently there has been a trend toward the application of pre-filled syringes, but typically these are developed once the product has proven itself in early clinical testing.
Conclusions
Clearly many challenges must be overcome to develop and scale-up a research discovery of a potential new therapeutic into a biopharmaceutical. Through systematic application of sound principles for process engineering and protein science, as well as analytical and quality control and assurance practices, these challenges can be overcome to reach the goal of filing an accepted IND with regulatory authorities. Achieving this essential milestone is the first step toward determining if the potential biopharmaceutical is safe and efficacious.
If so, the rewards can be great for not only the product’s developers but also for the patients and their healthcare providers.
References
- See Pfenex corporation, www.pfenex.com
- See Ajinomoto Althea corporation, www.corynex.com
- S.R. Hamilton et. al., Humanization of Yeast to Produce Complex Terminally Sialylated Glycoproteins, Science, 313: 1441-1443 (2006).
- K.P. Jayapal, K.F. Wlaschin, & W.-S. Hu, Recombinant protein therapeutics from CHO Cells – 20 years and counting, Amer Inst. Chem Eng, CHO Symposium, Oct. 2007
- D. Ghaderi et. al., Production platforms for biotherapeutic glycoproteins. Occurrence, impact and challenges of non-human sialylation. Biotech Gen Eng Rev 28:147-176 (2012).
- See for example Protein Sciences application of insect-cell technology for vaccine production.
- FDA Guidance, Points to Consider in the Characterization of Cell Lines Used to Produce Biologicals”, 1993, and Federal Register/Vol. 63, No. 182/Monday, September 21, 1998/ Notices.
- W. Kusser & B. Lee, Novel, efficient approach of CHO media optimization using ambr15 and DoE, ESACT Conference, Barcelona, 2015. Presentation available from Sartorius Stedim Biotech.
- S. D. Jones, T.C. Ransohoff, F. Castillo, F.J. Riske, & H.L. Levine, High Throughput Process Development Approaches for Biopharmaceuticals, Amer Pharma Review, 27 Mar 2015.
- R.G. Harrison, P.W. Todd, S.R. Rudge, & D.P. Petrides, Bioseparations Science and Engineering, Oxford University Press, New York, 2015.
- U. Gottschalk (ed), Process-Scale Purification of Antibodies, Wiley-Interscience, Hoboken, NJ, 2009.
- Guidance for Industry: Q5A Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin, FDA, 24 Sept 1998.
- Guideline on Virus Safety Evaluation of Biotechnological Investigational Medical Products, EMEA, 28 Jun 2006.
About the Author
Dr. Michiel “Mike” E. Ultee formed Ulteemit BioConsulting in October 2013 to offer expert consultation in the fields of process development and manufacture of protein therapeutics. With over 30 years experience in the development of biopharmaceuticals, from research through commercial manufacturing, Dr. Ultee offers both creative insight and a rich perspective into what it takes to turn a protein discovery into a therapeutic.
“The Road to the Biologic IND” is a special program created by Dr. Ultee for those in the research and discovery to understand better the steps and commitments required to successfully develop and manufacture an earlyphase biopharmaceutical. It has now been presented in ten cities across the county, often to capacity crowds.
Dr. Ultee has published a wide variety of papers of all aspects of bioprocessing. These include an in-depth review of antibody purification techniques for the Encyclopedia of Industrial Biotechnology, initially in 1999 and then for its second edition in 2010. Other papers have focused on the production and purification of challenging proteins, including fusion proteins and IgM antibodies. More recently he has presented and published multiple times on single-use applications in bioprocessing, as well as on flexibility in bioprocessing.
As the scientific co-founder and CSO of Laureate Biopharma, where he worked for many years, Dr. Ultee succeeded in developing dozens of proteins into new biopharmaceuticals for Laureate’s clients. Through these activities as well as his publications and presentations, he became well known, respected and widely connected in the industry.
When Gallus Biopharmaceuticals acquired Laureate on October 1, 2013, Dr. Ultee agreed to become their CSO on a part-time basis. This arrangement allowed him to continue to work on new biopharmaceutical challenges for Gallus’ clients, while also permitting him time to consult for others. A year later Patheon acquired Gallus and for an interim period he served as CSO for their Biologics division. Dr. Ultee left Patheon in December 2014 and is now consulting full time.