Introduction
Controlled release (CR) has been a subject of continued interest, and the pharma industry is taking a closer look into release profiles of drugs because of risk factors associated with early release (dose dumping) before being delivered to intended sites to achieve the desired efficacy, and to overcome toxicity and drug abuse issues. The latter has become more challenging, especially in controlled delivery of opioids in pain management.1-2 Nonetheless, the former one is also poorly understood because of complexity associated with the mechanism of controlled release from a particular dosage.3
In spite of those challenges, the industry is aiming to design CR formulations that will help maintain an optimal drug concentration in systematic circulation between minimum toxic concentration (MTC) and minimum effective concentration (MEC), thus alleviating the side effects of dosages.4 There are various approved controlled release drugs, and those include Anpec® SR; Cordilox® SR; Imdur Durules, Isoptin® SR; Monodur Durules®; Nuelin® SR; Sinemet® CR; Theo-Dur®; Adalat® OROS; Adalat® tablets, Agon SR; Ceclor® CD; Felodur ER; Keflor® CD; Kinidin Durules®; MS Contin®; Naprosyn® SR; Plendil® ER; Tenuate Dospan® among others.5
Oral dosages with desired optimal release characteristics to meet the narrow therapeutic windows usually contain polymers either in matrices or coated around multi-particulates. Thus, these dosage forms can also be differentiated in single-unit or multiple-unit systems, each having benefits and drawbacks. Functionalities of these polymers vary depending on their chemistries and physicochemical properties; and those include cellulosic, acrylic, polyvinyl acetate, polyvinyl pyrrolidone, or polyethylene oxide among others. As a consequence, identifying an appropriate polymer that meets the desired release profile of one drug candidate may not be suited for other drug candidates due, in part, to inherent different molecular properties of APIs and/or polymers which could in turn affect the intermolecular interactions and influence the controlling mechanism.6 Such polymeric devices could also be categorized as diffusion controlled monolithic, solvent activated (swelling or osmotically devices), chemically controlled (biodegradable) or externally triggered systems (pH, buffer, temperature).7Figure 1 illustrates the release mechanisms of pharmaceutical dosages, which could be relevant for the characteristics of different polymers.8 Thus, finding an appropriate polymer that meets such challenges is highly warranted.
 Figure 1. Illustrations of instant and controlled release curves (A), and the deviations caused by the factors leading to slower (d, e) or faster (a, b, c) release from a CR dosage (B).
Figure 1. Illustrations of instant and controlled release curves (A), and the deviations caused by the factors leading to slower (d, e) or faster (a, b, c) release from a CR dosage (B).Synthetic, semi-synthetic and natural polymers are available for controlled delivery of drugs.9-10 However, for CR coating purposes the number of polymers is quite limited as there are ethyl cellulose, two acrylics and polyvinyl acetate. Since polyvinyl acetate is the newest polymer offered as an aqueous dispersion, this manuscript will focus on this compound, mainly on its physico-chemical properties and applications in controlling the release profiles of model drugs. The importance of using the PVAc with special emphasis on extended release of high water soluble drugs will also be examined. The coating challenges associated with curing, dose dumping, migration of plasticizers, affinity of polymer with the tablet/pellet surfaces, robustness of coating polymer and processes will also be reviewed and addressed in light of development of extended release dosages.
Polyvinyl acetate polymer
Polyvinyl acetate (MW 450,000) is a hydrophobic polymer and is also referred to as PVAc. It is insoluble and does not strongly swell as other extended release polymers such as xanthan gum, guar gum or locust bean gum, and hydroxyalkylated or carboxyalkylated cellulosic excipients. PVAc, which is available as 30% dispersion comprising of 2.7% povidone K30 as a pore former and 0.3% sodium lauryl sulfate (SLS) as a stabilizer/wetting agent.11 The povidone plays an important role in releasing the drug molecules from insoluble PVAc films and SLS provides an advantage for spreading the polymer during coating, hence leading to homogeneous films. In addition, the self-sealing property of PVAc is also crucial to prevent instant release and avoid any dose dumping (Ensslin et al., 2009).12 All these attributes are critical in formulation of a tablet dosage, and thus, the selection of a polymer like PVAc is important to meet the requirements for extended release and to design a better pill for patient compliance. A polyvinyl acetate based directly compressible matrix polymer that contains 80% of PVAc and about 20% of PVP K30 as a pore former13 will not be subject of current discussion in the article.
Physico-chemical properties of PVAc dispersion
The colloidal dispersion is prepared by emulsion polymerization in presence of sodium lauryl sulfate to help to adjust the particle size and also to improve the stability of dispersion by electrostatic charging and building the required zeta potential. The PVP that acts as a protective colloid and pore forming agent also helps to increase the stability of dispersion and controls drug release when the polymer film is exposed to an aqueous solution. The particle size of the dispersion is about 170 nm, with a very narrow and unimodal distribution. The minimum film forming temperature (MFT) is about 18 °C which makes it ideal for coating at product temperature of about 30-35 °C without a plasticizer. The pH 4.5 of the dispersion provides additional advantages of enhancing the chemical stability of polymer by preventing degradation of polyvinyl acetate to polyvinyl alcohol.
This article will describe some of the important attributes of PVAc polymer including (a) ability to control the drugs to meet the desired target release profile, (b) requirement for curing (c) effects of pH and temperature (d) effects of hydrophilic polymers as pore formers on the release profile (e) effects of compression on the robustness and stability of this flexible coating polymer, and (f ) for taste masking of bitter drugs.
Results
A. Release profile of theophylline
Coating suspension compositions and conditions are shown in Table 1. Figure 2 illustrates the release of theophylline pellets (0.8-1.3 mm in diameter) coated with PVAc dispersions at 0.5 mg/cm2, 1 mg/cm2 and 2 mg/cm2. It is obvious that an increasing amount of polymer coating slowed the dissolution from the pellets.
Table 1. PVAc coating suspension compositions and conditions of theophylline pellets

 Figure 2. Dissolution profile of theophylline coated with PVAc at different weight gains.
Figure 2. Dissolution profile of theophylline coated with PVAc at different weight gains.B. Release profile of propranolol
Effects of pH and curing conditions
The effects of pHs and curing conditions were investigated to understand the release of propranolol from PVAc coated pellets. Figure 3A illustrates the release of propranolol from pellets at pH 1.2, 4.5, 6.8 and 7.4, and Figure 3B illustrates the impact of different curing conditions such as 2 hours and 24 hours at 60 °C on drug release. The data clearly demonstrates that release was approximately 75% in 8 hours independent of pH and curing conditions.
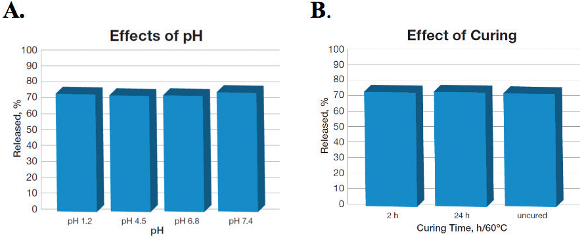 Figure 3. Propranolol release in 8 hours from PVAc coated pellets: (A: effects of pH and (B) effects of curing conditions (adapted from ref. Kolter et al., 2013).14
Figure 3. Propranolol release in 8 hours from PVAc coated pellets: (A: effects of pH and (B) effects of curing conditions (adapted from ref. Kolter et al., 2013).14Effects of plasticizers and talc
The effects of plasticizers and talc on the release profile of propranolol pellets coated with PVAc (Figure 4A) and talc (Figure 4B) were identical, wherein, the release ranged between 74% and 78% with and without plasticizers or talc. The release was also independent upon the type of plasticizers used, as shown in Figure 4A.
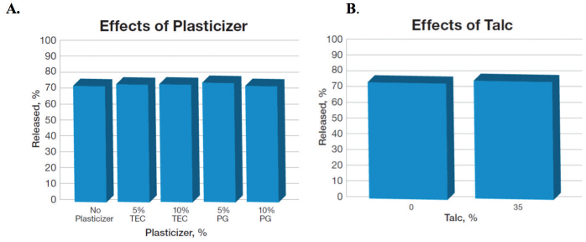 Figure 4. Effects of plasticizers and talc on propranolol release in 8 hours from the pellets coated with PVAc at 15% weight gain (adapted from ref. Dashevsky et al., 2005).15
Figure 4. Effects of plasticizers and talc on propranolol release in 8 hours from the pellets coated with PVAc at 15% weight gain (adapted from ref. Dashevsky et al., 2005).15Effects of compression force
The effect of compression force on propranolol tablets prepared by tabletting of pellets coated with PVAc was investigated and compared with a cellulosic excipient. Figure 5 illustrates that the compression was more imminent on the cellulosic based excipient as compared to PVAc films containing 25% (cellulosic) and 10% (in PVAc) of TEC as a plasticizer. In PVAc coating, for example, propranolol release was almost independent of the compression force, while with ethyl cellulose it increased as a function of compression force from 5 kN to 25 kN, attributed primarily due to fracture of the cellulosic films, hence causing the faster release, as shown in Figure 5A.
 Figure 5. Influence of compression force on propranolol release in 8 hours from tablet derived from (A) ECD/25% TEC and (B) PVAc/10% TEC coated pellets and compressed at different compression force (coating level 20% and pellet content 50%; adapted from Dashevsky et al., 2004).16
Figure 5. Influence of compression force on propranolol release in 8 hours from tablet derived from (A) ECD/25% TEC and (B) PVAc/10% TEC coated pellets and compressed at different compression force (coating level 20% and pellet content 50%; adapted from Dashevsky et al., 2004).16Effects of granule size and API level
The effect of granule size on API’s release was studied on the PVAc coated propranolol pellets and tablets compressed from the granules varying in particle sizes, and the release profile was compared with those corresponding tablets derived from the same granules. For example, the drug release of the smaller (500-600 and 600-710 microns) or larger granules (710-850 microns and 850-1000 microns) were similar at all conditions investigated (e.g. approximately 75% in over 8 hours), suggesting that the release was independent upon the pellet size and/or compressed tablets from the corresponding granules. It was also apparent with selection of different fillers/binders in the PVAc coated propranolol tablets compressed at 15 kN, wherein, the release remained about 75% in 8 hours, and was within the range regardless of the excipient composition used as a filler.
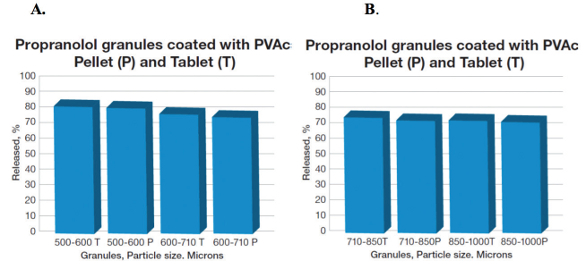 Figure 6. Propranolol release in 8 hours from PVAc/10% TEC coated pellets (and compressed tablets at 15 kN) with different granule sizes; A: 500-600 and 600-710 microns; and B: 710-850 microns, and 850-1000 microns (coating level 20%, and pellet content 50% (Adapted from ref. Dashevsky et al., 2004).16
Figure 6. Propranolol release in 8 hours from PVAc/10% TEC coated pellets (and compressed tablets at 15 kN) with different granule sizes; A: 500-600 and 600-710 microns; and B: 710-850 microns, and 850-1000 microns (coating level 20%, and pellet content 50% (Adapted from ref. Dashevsky et al., 2004).16PVAc coated pellets compressed into tablets with varying amounts of pellets at a compression force of 15 kN, and release of propranolol were also investigated. Figure 7 illustrates the release data from the tablets prepared with 25%, 50%, 75% and 90% of pellets and with corresponding amounts of microcrystalline cellulose as a filler. The data clearly suggests that increasing amounts of pellets in tablets did not significantly influence the release profile of propranolol from the tablets. Principally, it means that the pellets are so deformable and plastic in their compression behavior that they do not require large amounts of a plastic binder to protect them.
 Figure 7. Drug release in 12 hours from PVAc/10% TEC coated (20% wt. gain) pellets and tablets compressed with microcrystalline cellulose as a filler (Adapted from Dashevsky et al. 2004).16
Figure 7. Drug release in 12 hours from PVAc/10% TEC coated (20% wt. gain) pellets and tablets compressed with microcrystalline cellulose as a filler (Adapted from Dashevsky et al. 2004).16C. Effects of a pore former
Figure 8 illustrates the effects of a hydrophilic graft PEG-PVA copolymer on release profile of propranolol tablets over 8 hours. The figure reveals that PVAc is extremely effective in retarding the dissolution at 16 mg/cm2 coating level, but as the amounts of PEG-PVA increase, the film allows more water to penetrate resulting in greater release of drug through the PVAc film, and at a 1:1 ratio, the drug was released completely in 8 hours. This can be taken to suggest that the hydrophilic polymers like PEG-PVA, and others, can be used to adjust the release profile of a controlled release dosage even when large coating thicknesses are applied. In this example thick coatings were used to completely prevent dose-dumping, because coated single unit drug delivery system (DDS) are more prone to this effect than multiple-unit DDS.
 Figure 8. Effects of PEG-PVA as a pore former on propranolol release from PVAc coated tablets (adapted from ref. Kolter et al., 2013).14
Figure 8. Effects of PEG-PVA as a pore former on propranolol release from PVAc coated tablets (adapted from ref. Kolter et al., 2013).14D. Taste-masking of acetaminophen
PVAc has been evaluated in taste-masking of a number of bitter drugs including ibuprofen, acetaminophen and others. Table 2 shows the formulation compositions and coating conditions of acetaminophen pellets, and Figure 9 illustrates the release profile of taste masked drug.
Table 2. Coating condition of acetaminophen
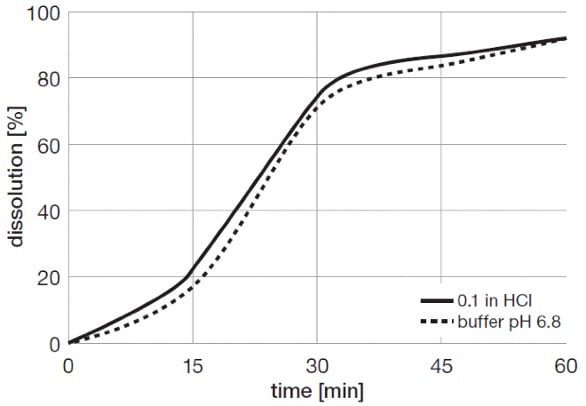 Figure 9. Release profile of taste-masked acetaminophen pellets coated with PVAc
Figure 9. Release profile of taste-masked acetaminophen pellets coated with PVAcDiscussion
Huang and Brazel suggested a few options to control the initial burst of drugs from a matrix that include (i) by extraction of surface bound drugs, (ii) by surface modification and (iii) applying an outer coating of API’s with varied polymer compositions.17 The latter is widely practiced today in industry because of its simplicity and also availability of different pharmaceutical polymers suited for the desired controlled release profiles.
Understanding the drug release phenomenon
Diffusion, degradation and swelling are primary mechanisms of drug release from the controlled delivery systems. In a diffusion process, the drug moves between the polymeric chains and out to the aqueous media. The movement of drug molecules depends upon the type of matrix and also the other components that could expedite it by increasing the permeation of water due to hydrophilic components in the matrix or slow it down by gelling of the matrix. In PVAc coating, for instance, the presence of a hydrophilic component povidone K30 facilitates the release as it dissolves by allowing water to penetrate into the films surrounding the pellet or tablet. As a consequence, the diffusion rate of drug remains fairly stable over the lifetime of delivery system until drug dissolves and releases out completely through the polymeric membrane. Thus, controlling the delivery that meets the therapeutic index will also further reduce toxicity of drugs.18-19
Kibria et al. investigated the coating of salbutamol sulfate (SS) pellets with PVAc and compared with cellulosic and acrylic polymers and found that the release profile of SS was well controlled with PVAc as opposed to other polymers, that is, only 26% of drug was released in the first hour and release was continued to increase as the time progressed and completed in 10 hours.20 The authors concluded that the sustained release of SS was attributed to smooth and uniform coating of pellet surface. In PVAc coating with propranolol pellets, the release (t50 released in 6 hours and t75 in 8 hours) was well controlled and robust, and was also nearly independent of compression force in case of tableting of pellets (Figure 5B). In contrast with the cellulosic excipient dissolution was faster as the compression increased, suggesting that the films might have been ruptured during compression (Figure 5A).21 For attaining a certain release profile with PVAc, 3 major factors must be taken into consideration: aqueous solubility of the drug, the coating level and the amount of hydrophilic pore former. In a recent study, ascorbic acid pellets coated with 5% PVAc showed about 70% release in over 6 hours which matched the dissolution profile of a commercially available CR reference product.22

Shao et al. investigated the effects of temperature and plasticizers on PVAc coated diphenhydramine hydrochloride nonpareil beads, and observed that the API release was dependent upon the type of plasticizers and the storage conditions. Increasing the temperature to 80 °C stabilized the API release from the beads containing 15% propylene glycol as a plasticizer, suggesting that some level of curing at higher temperature is necessary.23 The effect of plasticizers was also noteworthy in processing and authors conclude that the plasticizers should be evaluated in PVAc coatings in the early stages of formulation development. In our study, however, we observed that the curing effect of propranolol pellets coated with PVAc and 10% TEC as a plasticizer was insignificant at 2 hours and 24 hours of storage conditions at 60 °C.14
Several factors are responsible for retarding or expediting the release of drugs from controlled release excipients including the inherent physico-chemical properties of polymers such as pH/ionic buffer, glass transition temperature, and minimum film forming temperature. For instance, polymers with higher MFT (or Tg), require higher amounts of plasticizers in coating to reduce the MFT. Typically, a temperature of 10-15 °C above the MFT is required for coalescence of droplets to yield a smooth coating. Thus, for PVAc with an MFT of 18 °C a typical processing temperature should be around 30-35 °C. In contrast, acrylic and cellulosic polymers with MFT values typically ranging between 39 °C and 81 °C required higher percentages of plasticizers for uniform coatings, and curing was also essential depending upon the polymer type and the amount of plasticizers. In contrast, an acrylic polymer with lower MFTs (ca. 5 °C), shows a rapid coalescence of droplets, but typically also result in tackiness of the films. In such cases, an anti-tacking agent is required to alleviate the tackiness, and manage the coating process. In PVAc based coatings, however, the data suggests that anti-tacking agents such as talc did not impact on the release profile of propranolol pellets (Figure 4B). In addition, the release profile of propranolol from pellets was independent upon the plasticizers’ type (propylene glycol and triethyl citrate) and was identical to pellets lacking any plasticizers (Dashevsky et al. 2005).15
Active ingredients (APIs) may have tendencies to migrate into coatings, resulting in tackiness and/or faster dissolution. For instance, ibuprofen in PVAc and cellulosic polymers showed the migration or diffusion and crystallization of drug within and onto the film surface, causing to dissolve relatively faster than anticipated. The sub-coating with 5% PVA, however, prevented the migration and stabilized the release profiles for over 6 months at 40 °C. Other contributing factors such as inclusion of anionic surfactants in coating solution of cellulosic polymers, and adjusting pH/ionic buffer for acrylic polymers resulted in retardation of release from the pellets.24-25 The lag time of a metformin gastroretentive (GR) tablet was also dependent upon the coating polymers. For example, the GR dosages coated with PVAc showed slightly longer floating time as compared to coated tablets with an acrylic base polymer, but the release profiles were similar under identical conditions and well controlled with increasing coating levels between 4 mg/cm2 and 10 mg/cm2 as opposed to the acrylic polymer. The effects of paddle speed and pHs on dissolution of GR tablets were less significant on the release with PVAc coated tablets as compared with those coated with acrylic polymer (Eisenächer et al. 2014). 26
Conclusion
This study highlights the characteristics of PVAc in controlled release formulation of pellets, compressed pellets and coated tablets. Apparently, the release profile for a certain drug can easily be adjusted by 2 major parameters, which are coating level and ratio of PVAc to hydrophilic polymer. The latter acts as a pore former increasing the release rate. PEG-PVA copolymer has been proven to have very beneficial properties in combination with PVAc because it is the most flexible water-soluble pharmaceutical polymer and it is highly compatible with PVAc. The high flexibility of PVAc indicated by the low Tg and high elongation at break makes this polymer, in combination with PEG-PVA copolymer or without, an ideal tool for compressed pellets since they do not suffer from fractures upon compression and subsequently from a burst release. A coating level of about 2-5 mg/ cm2 for pellets seems to be appropriate since it enables complete and homogeneous coating with an economical coating process. The low minimum film-forming temperature (MFT) allows coating formulations without plasticizers but plasticizers might be beneficial for the film homogeneity and curing effects. Depending on the coating recipe, the coating parameters especially the product temperature and the drying step in the coater, curing might be necessary.
An innovative coated single unit DDS consisting of a regular tablet core and a coating based on PVAc and PEG-PVA at a coating level of 10 – 20 mg/cm2 can serve as a substitute for the complex OROS system, since it generates similar release profiles. The relatively thick coating is required in order to eliminate the risk of dose-dumping and to assure that each DDS releases the drug in the same way. In addition, PVAc can also be used for taste-masking of bitter APIs, particularly when it is combined with large amounts of PEG-PVA. In this case the hydrophilic copolymer enables a quick release and prevents retardation. PVAc, especially in the form of a 30% aqueous dispersion has excellent physico-chemical and coating properties facilitating robust and reliable sustained release coatings, simple and quick product developments including release adjustments and economical processing, The unique self-healing properties of PVAc film-coatings support robustness of such DDS since damaged parts are sealed again preventing a dose-dumping effect or a significant change of the release profile (Ensslin et al., 2009).12
PVAc dispersion has been used in small quantities (approx. 5%) as a granulating aid in manufacturing of mannitol based orally dispersible excipient to help increase the compressibility of granules.27 It has been marketed in several orally dispersing tablets (ODTs). PVAc dispersion is monographed in the United States and European pharmacopeias and has been approved in many drug products. The safety of PVAc polymer has been documented in literature as it is widely used in chewing gum applications.
References
- J. Bartholomäus, S. Schweir, M. Brett, H. J. Stahlberg, E. Galia, and K. Strothmann, Abuse deterrent technology: New abuse deterrent formulation (ADF) technology for immediate release opioids, Drug Dev. Delivery, 2013, 13, 76-81.
- L. Ma, L. Deng, J. and Chen, Applications of poly(ethylene oxide) in controlled release tablet systems: a review, Drug Dev Ind. Pharm. 2014, 40, 845-851.
- H. Ahmad, I. Khalifeh, B. Alkhaldi, K. Aiedeh, and H. S. Alkhatib, Application of active layering and coating techniques in the development of multiparticulate, controlled release dosage form of a high-dose, highly soluble drug, Pharm. Dev. & Tech., Early online, 1-9; DO I:10.3109/10837450.2013.805778.
- R. A. Siegel and M. J. Rathbone, Overview of controlled release mechanism, In Fundamentals and applications of controlled release drug delivery, Eds. J. Siepmann et al., Chapter 2, 19-43, Springer 2012.
- L. N. Sansom, Oral extended release products, Aust. Prescr., 1999, 22, 88-90.
- G. Vilar, J. T-Puche and F. Albeicio, Polymers and drug delivery systems, Current Drug Del., 2012, 9, 1-28.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Polymers for drug delivery systems, Annu. Rev. Chem. Biomed Eng., 2010, 1, 149-173.
- P. Gao, X. Nie, M. Zou, Y. Shi and G. Cheng, Recent advances in materials for extended release antibiotic delivery system, The J. Antibiotics, 2011, 64, 625-634.
- J. R. Joshi and R. P. Patel, Role of biodegradable polymers in drug delivery, Int. J. Current Pharm. Res., 2012, 4, 74-81.
- R. Gwozdz, Polymers for solid oral dosage forms. Drug Development & Delivery, 2012, 12, 34-37.
- Kollicoat® Grades: Functional polymers for the pharmaceutical industry, Editor V. Buhler, BASF January 2007; (a) Chapter 4: Kollicoat® SR 30D - Polyvinyl acetate dispersion 30%; Technical brochure (BASF) (b) Chapter 2: Kollicoat® IR - Polyethylene-Polyvinyl alcohol graft copolymer, Technical brochure (BASF).
- S. Ensslin, K.P. Moll, T.Haefele-Racin and K. Mäder, Safety and robustness of coated pellets: self-healing film properties and storage stability, Pharm. Res. 2009, 26, 1534-1543.
- Kollidon® Textbook, Editor: V. Buhler; 9th Edition, Chapter 4; Kollidon® SR- Polyvinyl acetate powder for matrix, Technical brochure (BASF).
- K. Kolter, A. Dashevsky, M. Irfan and R. Bodmeier, Polyvinylacetate based film coatings, Int. J. Pharm., 2013 457, 470-479.
- A. Dashevsky, K. Wagner, K. Kolter, and R. Bodmeier, Physicochemical and release properties of pellets coated with Kollicoat® SR30D, a new aqueous polyvinylacetate dispersion for extended release, Int. J. Pharm., 2005, 290, 15-23.
- a) A. Dashevsky, K. Kolter and R. Bodmeier, Compression of pellets coated with various aqueous polymer dispersions, Int. J. Pharm. 2004, 279, 19-26; (b) A. Dashevsky, K. Kolter, and R. Bodmeier, pH independent release of a basic drug from pellets coated with the extended release polymer dispersion Kollicoat® SR 30D and the enteric polymer dispersion Kollicoat® MAE30DP, Eur. J. Pharm. Biopharm., 2004, 58, 45-49.
- X. Huang and C. S. Brazel, On the importance and mechanisms of burst release in matrix controlled drug delivery systems, J. Control. Rel., 2001, 73, 121-136.
- L. Brannon-Pappas, Polymers in controlled drug delivery, 1997, www.mddonline.com
- H. P. James, R. John, A. Alex and K. R. Anoop, Smart polymers for the controlled delivery of drugs-a concise overview, Acta Pharmaceutica Sinica B, 2014, 4, 120-127.
- G. Kibria, M. S. Islam, and R. Jalil, Comparative study of aqueous dispersion of different types of polymers on release behavior of salbutamol sulphate from coated pellets, J. Pharm. Sci Res., 2010, 2, 107-113.
- P. Bansal, S. Vasireddy, and D. Parikh, Effect of compression on the release properties of polymer coated niacin granules, J. Control. Rel. 1993, 27, 157-163.
- F. Andreazza and H. G. Ferraz, Preparation of pellets containing high soluble drug by extrusion/spheronization and coating with Kollicoat® SR 30D, Braz. Arch. Biol. Technol., 2011, 54, 315-320.
- Z. J. Shao, L. Morales, S. Diaz, and N. A. Muhammad, Drug release from Kollicoat® SR30D coated nonpareil beads: Evaluation of coated level, plasticizer type, and curing condition, AAPS PharmSciTech, 2002, 3(2), 1-10, article 15.
- R. Bodmeier and O. Paeratakul, Process and formulation variables affecting the drug release from chlorpheniramine maleate-loaded beads coated with commercial and self-prepared aqueous ethyl cellulose pseudolatexes, Int. J. Pharm. 1991, 70, 59-68.
- R. Bodmeier, X. Guo, R. E. Sarabia, and P. F. Skultety, The influence of buffer species and strength on diltiazem HCl release from beads coated with the aqueous cationic polymer dispersions, Eudragit® RS, RL 30D, Pharm. Res., 1996, 13, 52-56.
- F. Eisenacher, G. Garbacz, and K. Mader, Physiolgical relevant in vitro evaluation of polymer coats for gastroretentive floating tablets, Eur. J. Pharm. Biopharm., 2014, 88, 778-786.
- S. Ali and K. Kolter, A recent advancement in orally disintegrating formulations, Am. Pharm. Rev., 2014, 17, 1-6 (May/June).
Author Biographies
Shaukat Ali has over 21 years of experience in the pharmaceutical industries including 11 years at BASF, where he supports solubilization, instant and modified release platforms, and APIs. Dr. Ali’s areas of expertise include solid dispersions, liposome drug delivery, controlled release, transdermal, and film development technologies. Dr. Ali is a member of the editorial advisory boards of American Pharmaceutical Review, Biopharma Asia, Contract Pharma, Drug Delivery & Development, International Journal of Pharmaceutical Investigation, Journal of Pharmaceutical Science and Pharmacology, and Journal of Analytical & Pharmaceutical Research. He is also USP panel of experts for General Chapters-Physical Analysis. He received his Ph.D. in Chemistry from the City University of New York and pursued his postdoctoral interest at the University of Minnesota and Cornell University. He has authored over 37 scientific articles and is co-inventor in 14 US patents.
Karl Kolter, after having worked for 7 years at Knoll AG in Ludwigshafen, joined BASF AG in 1993, where he has been responsible for R&D activities in pharmaceutical excipients, drug formulations and the application technology of vitamins and carotenoids for pharma and food. Dr. Kolter’s current work is the development of innovative excipients mainly for solid oral dosage forms, which has already resulted in various new products in the Kollicoat® and Kollidon® range (Kollicoat® MAE 30 D, 100P, SR 30 D, IR, IR White, Protect, Kollidon® SR, CL-F, CL-SF, Ludipress® LCE). He obtained his Ph. D. in pharmaceutical chemistry at the University of Mainz (Germany). He has published more than 100 articles and posters, and is inventor in more than 90 patents.
Bernhard Fussnegger has over 25 years of experience in the excipients business of BASF. Presently he is in charge of product development activities as well as the instant and modified release platform. In this area, he is responsible for technical aspects of excipients manufactured internally and externally of BASF. Dr. Fussnegger is a member of several expert panels including the chair of the USP expert panel on Povidones, vice-chair of the pharmacopoeial review and harmonization committee of IPEC Europe, and he is delegate of IPEC Europe in the IPEC/PDG discussions on harmonization efforts for excipients. He received his PhD in food chemistry from the Technical University of Stuttgart. He has authored several scientific articles and is inventor in more than 20 patents.