Introduction
Quality by Design (QbD) is a systemic approach to pharmaceutical development. It is essentially the information gathered through various stages of designing and developing formulations and manufacturing to provide scientific understanding and to support the design spaces for specification and manufacturing controls. This includes defining Quality Target Product Profile (QTPP) and designing product manufacturing processes; identifying Critical Quality Attributes (CQA), Critical Process Parameters (CPP), Critical Material Attributes (CMA) and the sources of variability; controlling manufacturing processes to ensure the product meeting predefined quality consistently.
Nasal suspension manufacturing and primary packaging process (filling into a bottle and further assembling with a metered nasal pump and an actuator with an over-cap) is a complex process. The QTPP for a nasal suspension is to be in-vivo and in-vitro bioequivalence and bioavailability to the corresponding reference listed product, which is a critical milestone in the product development process. Once this milestone is achieved, further challenges are focused on reproducibility from the pilot scale to the commercial scale of manufacturing. To ensure success of the clinical bioequivalence batches and to gain thorough understanding of the manufacturing process, multivariate studies based on QbD principles should be conducted at various stages of product development from formulation development to pilot scale, such QbD studies must be conducted during the scale-up to the commercial scale.
This article briefly discusses the basis of formulation development and primary packaging selection, with a focus on the scale-up and qualification of a robust manufacturing process for a nasal suspension drug product following the QbD concept.
Formulation Development
The primary objectives in developing a nasal suspension1,2 include stability, compatibility, drug delivery characteristics and the good knowledge of the characterization of the active pharmaceutical ingredient (API).
In addition to fully understanding the physiochemical properties of the API such as crystallinity, polymorphism, flow characteristics, particle size, and chemical stability, it is important to evaluate the suitable suspension vehicle, which is critical to the formulation, since it influences the drug delivery and the QTPP. Typical viscosity imparting agents include natural polymers such as gellan gum, xanthan gum, guar gum and cellulose derivatives such as Avicel® (different grades available), Hydroxymethyl cellulose, Hypromellose etc. Special attention is required in selection of buffer agents, surfactant/dispersant (critical for product settling and resuspendability) and preservative system in terms of securing the product shelf-life stability.
Multivariate studies are usually conducted and many different physical and chemical factors are adjusted to achieve a quality bulk formulation. For a nasal suspension one of the most challenging studies includes optimization of the stable drug product with the primary packaging components also known as container closure system.
Container Closure System
 Figure 1. Illustration of a typical container closure system for a nasal product
Figure 1. Illustration of a typical container closure system for a nasal productUnlike oral dosage products, nasal spray product performance is governed by the delivery device, a container closure system as shown in Figure 1.
The container is generally HDPE or glass bottle crimp-able/screw-able with a metering nasal pump and a plastic actuator with an over cap. Selection of the suitable combination of the nasal pump assemble is critical to meet established QTPP elements such as in-vivo efficacy, invitro bio equivalency and bioavailability as specified in the FDA nasal product guidance as well as their compatibility with the product2.
Manufacturing Process Development
The following objectives from the regulatory guidance3 are applied at various scales (from pilot scale to commercial scale) during scale-up manufacturing process development.
- Objective 1: Identify critical material attributes (CMA) and critical process parameters (CPP) that have direct impact to the process and the performance of finished product
- Objective 2: Design and conduct experiments to establish suitable parameter ranges when appropriate
- Objective 3: Analyze the experimental data statistically to determine the criticality of the parameters
- Objective 4: Develop a control strategy for levels and ranges of the critical attributes and parameters
Identification of Critical Material Attributes and Process Parameters
The good understanding of the drug substance and critical excipient is generally achieved through bench top and pilot scale studies3. For a nasal suspension product, viscosity is a critical parameter used in the determination of robustness of the process through product shelf life. Therefore when a viscosity imparting agent such as cellulose is used, a proper hydration of the cellulose and the ability to withstand manufacturing conditions must be evaluated. Equally important, the API dispersion process and its uniformity in the bulk suspension should be investigated thoroughly.
The important step in determining a successful scale-up is to perform a thorough and in-depth risk assessment of the manufacturing process/equipment stepwise to understand gaps and criticality of the process through identified CMA. Many of the gaps can be eliminated based on engineering solutions and some of the critical steps can be assessed or mitigated through product development studies at various scales of manufacturing. There are many different types of risk assessment tools3 based on ICH Q9 guidance that can be employed to perform risk/gap analysis for the selected manufacturing process. The most commonly used tool is the Failure Mode and Effect Analysis (FMEA). The FMEA approach helps to understanding inherent limitations of the process and how the controls can be put in place to either monitor them or develop engineering solution to eliminate them. Utilizing the knowledge gathered from similar suspension products helps immensely in reducing the process gaps.
Figure 2 is a quick summation of both formulation and process risks specific to the compounding stage represented as heat mapping. The heat mapping is a high level overview of the risks identified from the FMEA, which was employed for the compounding stage in relation to formulation, process and equipment. Another approach that can be used is the fishbone diagram – “cause and effect analysis” to evaluate the risks4.
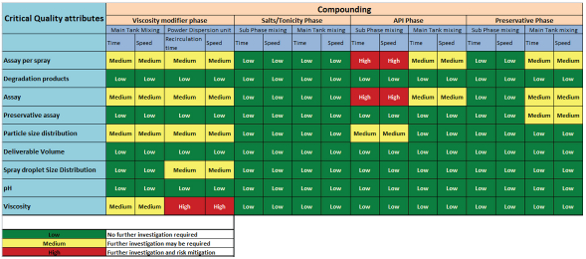 Figure 2. Risk assessment representation through heat mapping.
Figure 2. Risk assessment representation through heat mapping.After the risk assessment is completed, corresponding studies to further investigate and mitigate risks associated with the product quality should be conducted. The outcome of these studies generally includes determination of batch size suitable for commercialization, equipment operating parameters that can impact in-process and finished product Critical Quality Attributes (CQA) and CPP for container closure system.
Determination of Targets and Ranges for High Risk Parameters and Attributes
After the risk assessment for the parameters that have the most influence on the quality of the product, the next step is to determine the design space, ranges and targets for identified critical process parameters5.
To assess the impact of the variation of the CPP to the product, a series of Design of Experiments (DoEs) should be conducted to evaluate the effect of excipients utilizing the placebo and API containing batches. The purpose of these experiments is to evaluate the influence of the critical excipients on the product performance, such as spray pattern, droplet size, the API particle size distribution and assay per spray.
Factors to be evaluated include varying amounts of critical excipients, order of addition and cleaning efficacy. These studies establish the specifications for the in-process parameters, and provide an understanding of the relationship between the process/ equipment critical parameters and the product CQAs. For an example, Figures 3 to 5 show that the shear rate/mixing speed in combination with the time has significant impact on the finished bulk, where the viscosity value is directly linked to the mixing parameters. These figures reflect a typical suspension product with a thixotropic fluid nature and a non-Newtonian property. Figure 6 represents the finished bulk product studies at static, re-mixing and continuously mixing. Under static conditions the finished bulk tends to show upward trends in viscosity until it reaches a level of stabilization. Such studies provide valuable information in setting optimal specification ranges for CQAs.
 Figure 3. Product viscosity in relation to mixing speed and shear rate
Figure 3. Product viscosity in relation to mixing speed and shear rate Figure 4. Product viscosity in relation to mixing time
Figure 4. Product viscosity in relation to mixing time Figure 5. Product viscosity in relation to standing time (static condition) of finished bulk
Figure 5. Product viscosity in relation to standing time (static condition) of finished bulk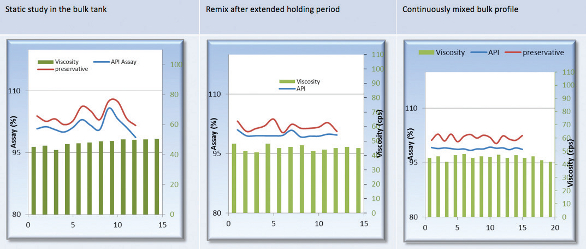 Figure 6. Finished bulk product studies under different conditions
Figure 6. Finished bulk product studies under different conditionsStatistical Analysis
Evaluation and analysis of the data obtained through multiple experiments is needed for organization of coherent data sets and drawing inferences from them before finalizing the process steps5,6. A good statistical analysis provides a solid foundation to develop effective control strategies for process variability and process capability at various stages of manufacturing process as well as provides valuable information on how the variations impact the acceptance criteria of the quality attributes7,8. In a recent publication by Apotex9 an improved and innovative statistical approach for probability acceptance (Pa) that provides a scientifically unbiased analysis towards how variation impacts the likelihood in a manufacturing process is described.
Figure 7 is a typical example of process capability statistical calculations using assay of drug substance (one of CQAs) to establish and control critical manufacturing process parameters10.
 Figure 7. Process capability of assay of API
Figure 7. Process capability of assay of APIThe two pie charts in Figure 8 are depiction of how variations in assay of API and preservative that were used as homogeneity indicators in determination of potential sources influencing the variability. Such analysis is important to control the critical parameters that have the most influence to cause variability in the process.
 Figure 8. Assay and preservative total variance (%)
Figure 8. Assay and preservative total variance (%)Process Control Strategy
After conducting all the process development studies and determining the critical process parameters, manufacturing process controls should be established and implemented. This ensures the consistency between batches and overall robustness of the product through its life cycle.
Effectiveness of process controls is demonstrated through in-process tests and checks through various manufacturing stages in a conventional process7,8. Use of simple Process Analytical Technology (PAT) such as in-line viscometers, can serve as homogeneity monitors whereas advanced technologies such as Focused Beam Reflectance Measurement (FBRM) is becoming a popular procedure for measuring the particle size in a suspension product. Real time monitoring through PAT is encouraged by many regulatory agencies across the globe. PAT helps considerably in reducing batch to batch variation and ensures product quality and safety. Table 1 below is a brief control strategy overview for a nasal suspension product.
Table 1. Control Strategy Overview
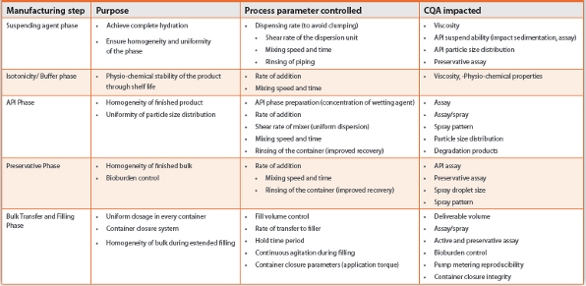
Conclusion
In the current regulatory landscape and complex manufacturing technologies, the product development of any dosage form has its own complexities and challenges. Nasal suspensions in particular need rigor for not only a robust manufacturing process but also device performance (spray pattern, drop size and assay/ spray…etc.) and compatibility of various components that have direct contact to the product.
Using QbD concepts and conducting series of designed experimentation based on the risk assessments of the QTPP, a successful commercial scale manufacturing process can be established for a given nasal suspension product. The product quality is assured by understanding and controlling the formulation and manufacturing variables. Performing statistical analysis for the data collected through the complex laboratory development, scale up and commercial manufacturing is critical to provide an assessment of the reliability of the process.
References:
- ICH Q8: Pharmaceutical Development. http://www.ich.org
- Center for Drug Evaluation and Research (CDER): Guidance for Industry Nasal Spray and Inhalation Solution, Suspension, and Spray Drug Products — Chemistry, Manufacturing, and Controls Documentation, July 2002. U.S. Food and Drug Administration. http://www.fda.gov
- ICH Q9: Quality Risk Management. http://www.ich.org
- Nancy R. Tague’s The Quality Toolbox, Second Edition, ASQ Quality Press, 2005
- Guidance for Industry: Process Validation: General Principles and Practices, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Center for Veterinary Medicine (CVM). http://www.fda.gov
- ICH Q10: Pharmaceutical Quality System. http://www.ich.org
- Current Good Manufacturing Practice for Finished Pharmaceuticals. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=211&showFR=1
- Guidance for Industry: Pharmaceutical quality of Inhalation and Nasal Products 2006 Health Canada. http://hc-sc.gc.ca
- Daniel Alsmeyer, Ajay Pazhayattil, Shu Chen, Francesco Munaretto, Maksuda Hye, and Pradeep Sanghvi: Acceptance Probability (Pa) Analysis for Process Validation Lifecycle Stages, AAPS PharmSciTech (# 2015)
- Montgomery D. Introduction to Statistical Quality Control. New York: Wiley; 2004. p. 776.
Author Biographies
Felix Francis is the Associate Director of Technical Operations – Liquid Dose at Apotex Inc. Toronto, Canada. He has over 17 years of experience in the pharmaceutical industry with focus on managing product life cycle from process development to commercial launch, process validation and further management of line / site transfers. Felix Francis holds a Master’s degree in Bio-chemistry and Microbiology from University of Mumbai, India.
Wan Jiang PhD is the Director Product Development-Liquid Dose at Apotex Inc. Toronto, Canada. She has over 25 years of working experiences in pharmaceutical industry at areas of formulation development, analytical and primary package development, stability program and manufacture process development for various finished product dosage forms including sterile products. Wan Jiang received a PhD. of Organic Chemistry in Iowa State University, USA.
Pradeep Sanghvi PhD is the Executive Vice President, Global R&D at Apotex Inc., Toronto, Canada. He has over 25 years of experience in pharmaceutical industry with strong technical, manufacturing, clinical, commercial, and regulatory knowledge of pharmaceutical dosage forms for global markets. He has pioneered several new technologies and managed the conceptualization, development and commercialization of brand and generic products. Pradeep Sanghvi holds over 50 worldwide patents and many publications in pharmaceutical technologies. He has a Bachelor’s degree in Pharmacy, Masters and Doctorate degree with specialization in Pharmaceutics.