The validation and implementation of rapid and alternative microbiological methods has gained significant momentum over the past decade, with multinational firms validating new technologies for a wide range of applications including finished product release testing (e.g., sterility), environmental monitoring, in-process control, Wfi analysis and microbial identification. When applicable, companies have submitted validation data to regulatory agencies and received approval to implement these same technologies for essential quality control uses. For example, Novartis obtained regulatory approval from more than 50 different countries for releasing their vaccine products using a rapid ATP bioluminescence sterility test.
More recently, stakeholders other than conventional pharmaceutical and biotech companies are beginning to embrace the need to utilize novel technologies that provide faster, automated and more sensitive microbiological results as compared with classical or conventional methods. Of note, the fast-growing gene and cellular therapy industry is looking toward rapid and alternative methods to demonstrate sterility for products that have a shelf-life much shorter than the required incubation time for the current compendial sterility test (e.g., 2-5 days vs. a 14-day incubation period). Additionally, compounding pharmacies that follow United States Pharmacopeia (USP) Chapter <797>, Pharmaceutical Compounding – Sterile Preparations,1 can release high-risk level compounded sterile preparations (CSPs) before receiving the results of a sterility test as long as there are written procedures requiring daily observation of the incubating test specimens and immediate recall of the dispensed CSPs in the event of any evidence of microbial growth in the test specimens. From a patient safety perspective, compounding pharmacies would benefit by using a validated rapid method that confirmed sterility prior to dispensing.
Over the past 15 years, the scientific community has utilized a number of guidance documents to demonstrate that rapid and alternative microbiological methods are suitable for their intended use. These include the Parenteral Drug Association (PDA) Technical Report Number 33, European Pharmacopoeia (Ph. Eur.) chapter 5.1.6 and United States Pharmacopeia (USP) chapter <1223>.
PDA Technical Report Number 33
The PDA published the very first validation guidance in its 2000 Technical Report Number 33 (TR33), Evaluation, Validation and Implementation of New Microbiological Testing Methods.2 The technical report offered guidance for evaluating, validating and implementing alternative and rapid microbiological methods to assure product quality. The original document also provided an overview of technologies and their applications. TR33 was significantly revised in 2013 under the new title, Evaluation, Validation and Implementation of Alternative and Rapid Microbiological Methods.3 The revised document highlights a number of topics, including, but limited to, a comparison of classical and new methods, the validation process, equipment and software qualification, method suitability testing, equivalence, the use of statistics, an updated technology overview, risk assessment, user requirements, implementation strategies and global regulatory expectations. Many firms have successfully utilized both the original and the revised versions of TR33 in support of their validation, regulatory submission and implementation endeavors.
Shortly after the publication of the original PDA TR33 (2000), the USP and Ph. Eur. published their own chapters on alternative microbiological method validation strategies. It should be noted that both of these chapters have been under a revision process for a number of years.
Ph. Eur. Chapter 5.1.6
Ph. Eur. chapter 5.1.6, Alternative Methods for Control of Microbiological Quality, was originally published in 2006.4 The chapter intended to facilitate the implementation and use of alternative microbiological methods where this can lead to efficient microbiological control and improved assurance for the quality of pharmaceutical products. Within the chapter, the basic principles for the qualification and validation of alternative methods were provided. In 2009, the European Directorate for the Quality of Medicines (EDQM) Alternative Microbiological Methods Working Party initiated a revision to the chapter. The result was a proposed draft that was published in the beginning of 2015.5
The scope of the proposed Ph. Eur. 5.1.6 revision focuses on the relevance of alternative methods to process analytical technology (PAT) concepts, improve the description of the methods and provide a clearer distinction between each, harmonize terminologies, better define user requirements, enhance the process of qualifying the instrument and the analytical method as well as improve the validation examples. As of the date of this publication, the period for soliciting public remarks ended and the Working Party began their review of the comments received.
Dr. Stephen J. Wicks, Regulatory Policy and Intelligence Officer (EDQM), recently provided an overview of the draft chapter during a 2015 USP workshop on alternative methods.6 Dr. Wicks stated the validation strategies proposed in revised chapter 5.1.6 are fairly aligned with the recommendations in the 2013 version of PDA TR33. It is hopeful that a final version of the chapter will be published in the near future.
USP Chapter <1223>
USP informational chapter <1223> Validation of Alternative Microbiological Methods, was originally published in 2006.7 In the summer of 2014, the USP published its proposed revision to the chapter in the Pharmacopeial Forum.8 Public comments were accepted until September 2014, at which time the USP expert committee worked on a final version. On June 1, 2015, the second supplement to USP38/ NF33 included the final version of chapter <1223> with an official date of December 1, 2015.9
The content of the revised USP chapter presents a significant modification from the original version published in 2006. The revision is intended to be less prescriptive and more flexible to accommodate all potential alternative microbiological methods, provide broader concepts relating to instrument and method validation and better define method suitability, user requirements, the use of statistical tools, non-inferiority concepts and equivalence models. For these reasons, the purpose of this paper is to summarize the new guidance provided in the new USP <1223> and to compare its recommendations with the concepts currently taught in PDA TR33 and the in-process revision of Ph. Eur. 5.1.6.
USP’s New User Requirements Section
The new chapter offers guidance on developing a user requirements specification (URS) that will identify the important functions and characteristics of the alternative technology including user interface, operational, environmental and space requirements. In developing the URS document, the end-user of the alternative method should consider the requirements for instrument qualification, the validation of the method (e.g., the alternative method should be equivalent to the existing method in terms of performance for its intended use) and method suitability (e.g., whether a sample or product will interfere with the outcome of the test).
PDA TR33 already includes a comprehensive section on user requirements and teaches the URS should describe the functions that the method and accompanying system must be capable of meeting that will be very specific for the end-user’s needs and the materials to be assessed. TR33 explains the requirements specified in the URS will directly influence the entire validation strategy and acceptance criteria and may well determine the success or failure of the method selection process by the end-user.
The in-process revision of Ph. Eur. 5.1.6 fundamentally mimics the recommendations provided in TR33 and suggests the URS at least addresses the application (e.g., qualitative, quantitative or identification), the level of sensitivity (limit of detection or quantification), microorganisms to be detected or identified (specificity), sample handling, time to detection and data management.
Both TR33 and Ph. Eur. 5.1.6 also provide guidance on when to perform a Design Qualification, which is a documented review and verification that the proposed design of the equipment or system is suitable for its intended purpose.
USP’s Discussion on the CFU and Alternate Signals
The colony-forming unit (CFU) is the unit of microbial enumeration in all current USP monographs. However, chapter <1223> describes the CFU as an estimation of cell counts with the potential for underestimating the true or actual number of microorganisms that may be present in a test sample. This may be due to a number of factors, including the physiological or stressed state of the microorganism(s), sampling technique and the ability of media and incubation conditions to allow the recovery and growth of microorganisms that may be present. Additionally, microorganisms existing in aggregates (e.g., clumps or chains) would normally grow into a single CFU, miscalculating the exact number of cells present. For these reasons, USP <1223> concludes the CFU cannot be considered the only unit of microbiological enumeration, and this concept introduces alternative signals or measurements from rapid and modern microbiological methods.
USP <1223> very briefly discusses the different signals from alternative technologies, such as direct cell counts from viability staining or autofluorescence methods. Although USP does not discuss technologies at length, both PDA TR33 and Ph. Eur. 5.1.6 provide an extensive review of the scientific principles and related signals for alternative methods that are based on microbial growth, metabolic activity or cellular components, viability staining, optical spectroscopy and nucleic acid amplification.
PDA TR33 also describes situations in which some of the alternative or rapid technologies provide a greater or improved level of detection sensitivity over its conventional method counterpart, either through design or the ability to detect stressed microorganisms. Furthermore, TR33 teaches in the event that an alternative or rapid method is qualified to provide greater sensitivity than the method intended to be replaced, an understanding of the impact to existing acceptance levels, in-process or product specifications, or compendia and regulatory expectations would be required. To support this matter, TR33 provides guidance on changing microbiological acceptance levels and specifications.
USP <1223> expands on this concept in relation to product performance and safety. The new chapter suggests that differences in an article’s observed cell count between an alternative signal and the classical CFU should not be a concern as long as the two methods used are equivalent to or are non-inferior to referee methods in terms of assessing the microbiological safety of the article. For example, if an alternative signal reports a higher cell count than the classical, growth-based CFU, this does not necessarily translate to a greater microbiological risk in the article, especially if the article has historically been shown to be safe and effective.
The CFU and Use of Statistics
USP <1223> states that attempts to use statistics to compare the CFU results to signals arising from biochemical, physiological, or genetic methods of analysis may have limited value because the different methods used cannot be expected to yield signals that could be compared statistically in terms of mean values and variability. Therefore, USP concludes the CFU cannot be used as acceptance criteria for the assessment of articles by alternative methods and it is the user’s responsibility to propose values (supported where necessary by scientific literature) that they can demonstrate are appropriate for the alternative method.
However, readers may be confused with this USP position for a number of reasons. As an initial matter, there are subsequent sections within USP <1223> that imply the CFU can be correlated with an alternative signal. For example, when demonstrating equivalency using Option 3 (“Results Equivalence” to the compendial method; see below), USP states to use a calibration curve showing a correlation between an alternative non-growth-based method and a growth-based method that reports outcomes in CFU. Next, under the “Equivalence Demonstration for Alternative Quantitative Microbiological Procedures” section (see below), USP advises one of the criteria for the verification of candidate alternative quantitative procedures is to show a high correlation between the results obtained from the candidate method and the compendial procedure. Therefore, the reader would understand a high correlation is taken to indicate that quantitative acceptance criteria expressed in CFU can be calibrated to criteria in the units of the alternative procedure. This is further supported by the USP’s recommendation in its 2014 in-process revision that stated, “it is generally possible to correlate [alternative method cell counts] with the estimates obtained using a compendial method.”8
Additionally, in the section describing how to demonstrate equivalence using correlation or linearity measurements, USP <1223> states, “the laboratory should determine a specification for the alternative method to correspond to the compendial specification for the required level of microbiological quality. For example, if the required level of microbiological quality is NMT 102 CFU, for which the compendial maximum acceptable count is 200 CFU, the laboratory will need to determine an acceptance criterion for the candidate alternative procedure that will match that value from the perspective of making a decision regarding microbial quality.” Therefore, the reader can interpret this to mean the CFU is an acceptable baseline measurement for which an alternative signal can be compared with.
Those versed in the art of rapid and alternative methods will also understand that PDA TR33 teaches the use of statistics to demonstrate equivalency or non-inferiority between an alternative and a growth-based compendial method. For example, TR33 states, “advances in technology can offer greater precision and sensitivity in comparison to conventional or compendial-referenced methodologies. As a result, an increase in organism recovery (i.e., detection or enumeration) may be observed. To address this, statistical treatment of the data generated from both methods should be used to demonstrate that the alternative or rapid method results are equivalent (i.e., noninferior) or better than (i.e., superior) that of the existing method.” This is more specifically defined in the requirement for accuracy, where quantitative cell counts are analyzed. TR33 states, “the new method should provide equivalent or better results than the existing method. The new method should provide a recovery of viable microorganisms not less than 70% of the actual recovery provided by the existing method for each suspension. Alternatively, a statistical comparison between the new method and the actual recoveries using the existing method may be performed.” In this instance, readers would appreciate the existing method refers to a growth-based procedure providing a CFU signal. TR33 further describes the statistical models that are appropriate for comparing the recovery in both methods: the Student t-test or Student t-test with Welch’s correction, a non-parametric test (e.g., Mann-Whitney-Wilcoxon or Kruskal-Wallis one-way analysis of variance test), an analysis of variance test with or without transformations, Post-tests (e.g., Tukey’s test) and other statistical equivalence or noninferiority tests. The appropriate test to use will depend on how the data are distributed and their variances.
The proposed draft of Ph. Eur. chapter 5.1.6 also recommends the use of statistical analyses when comparing the recovery of microorganisms using the growth-based compendial and an alternative method. For example, when discussing the accuracy criterion for the validation of an alternative quantitative test, chapter 5.1.6 states “[t] he alternative method should be shown to recover at least as many micro-organisms as the traditional method using appropriate statistical analysis.” In this case, accuracy of an alternative quantitative method is the closeness of the test results obtained by the alternative method to those obtained by the pharmacopoeial method. Similarly as described above, the reader would understand that the pharmacopoeial method would rely on the recovery of microorganisms in terms of a CFU.
Chapter 5.1.6 additionally provides three examples of a detailed protocol for the implementation of an alternative microbiology method. Under the example of a quantitative test for the enumeration of microorganisms, a solid-phase cytometry technology is specified, which is a non-growth-based method utilizing a viability stain and laser excitation. As a direct approach to demonstrating the equivalence of the two quantitative tests, both assays are performed side-by-side for a predetermined period of time (or number of samples). Chapter 5.1.6 specifies, “statistical analysis demonstrates that the results of the alternative method are at least equivalent to the pharmacopoeial method.”
USP permits the use of alternative methods to analyze compendial articles. Chapter <1223> states, “[a]lternative methods and/or procedures may be used if they provide advantages in terms of accuracy, sensitivity, precision, selectivity, or adaptability to automation or computerized data reduction, or in other special circumstances. If a product has proven safe in widespread use when released or controlled using current methods, the implementation of an alternative method which can be well-correlated to the existing method should be straightforward.” It would; therefore, make sense to employ the CFU as a baseline signal to demonstrate a correlation with an alternative method, especially since the CFU continues to serve as the exclusive unit of microbial enumeration in all USP monographs.
Finally, readers may not fully understand how they would necessarily propose values that they can demonstrate are appropriate for the alternative method in the absence of using the CFU as a baseline measurement to compare to. Although USP <1223> states this practice can be supported by scientific literature (see above), additional clarification may be warranted in a future revision of the chapter.
Understanding the USP’s Phases of Validation
USP <1223> explains alternative microbiological methods will generally provide a cell count signal or unit of measurement that is not a CFU. For this reason, the validation of such methods should involve the following phases:
- Qualification of the instrumentation or equipment
- Qualification of the analytical method using standardized microorganisms (in the absence of product)
- Demonstration of equivalence to the compendial method using standardized microorganisms (in the absence of product)
This is followed by method suitability, in which actual product or test samples are introduced.
Qualification of the Instrumentation
The first phase in validating an alternative method is to qualify the instrumentation or equipment that will be associated with the alternative method. USP <1223> directs the user to the guidance provided in USP <1058>, Analytical Instrument Qualification,10 and briefly lists the following phases to be addressed: the development of the URS, installation, operational and performance qualifications (IQ, OQ and PQ). PDA TR33 also features an adaptation to the USP <1058> model for the qualification of instrumentation and software, and provides a comprehensive discussion on how to conduct an IQ, OQ and PQ. The in-process revision of Ph. Eur. 5.1.6 also mentions IQ and OQ but provides an extended outline of how to perform a PQ. All three documents agree that the PQ is the phase of validation that should demonstrate the alternative method consistently performs in accordance with predetermined criteria (i.e., as specified in the user requirements) and thereby yields correct and appropriate results. Hence, the PQ can comprise qualifying the analytical method, demonstrating equivalence to the compendial or existing method and verifying the product or test sample does not significantly impact the performance of the alternative method.
Qualification of the Analytical Method
The next phase in USP’s validation design is to demonstrate the alternative method will detect or enumerate a relevant panel of microorganisms. During this phase of testing there is no comparison to the compendial method and the studies are conducted in the absence of actual product or test samples. Table 1 identifies the validation criteria that should be assessed for quantitative and qualitative methods and they are similar to what was advocated in the prior version of USP <1223>.
Table 1. Validation Parameters by Type of Microbiological Test
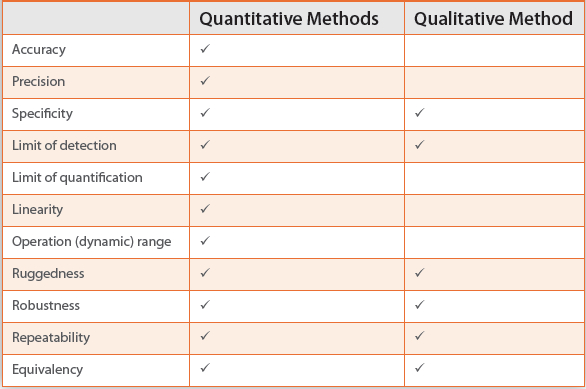
The approach summarized in Table 1 is generally comparable to the first phase of validation recommended in PDA TR33 and the proposed revision to Ph. Eur. 5.1.6. A few minor differences exist and these may be due to how each validation phase is identified and when the phase is performed (e.g., when a comparison to the compendial method is made). However, there is a significant difference between USP <1223> and the other two documents relating to the criterion “repeatability.” Repeatability is a subset of precision, and USP <1223> defines repeatability precision as follows: “The degree of agreement among individual test results when the procedure is applied repeatedly to multiple samplings of the same suspension of microorganisms and uses different suspensions across the range of the test. Also known as ‘repeatability’.” This definition is essentially identical to the definition of precision in PDA TR33 and very similar to what is defined in Ph. Eur. 5.1.6. However, precision (including repeatability) is not identified in either PDA TR33 or Ph. Eur. 5.1.6 as being a recommended validation parameter for a qualitative method.
Additionally, ruggedness does not appear in Ph. Eur. 5.1.6 as a validation criterion. This is not an issue because ruggedness (as described in the USP and TR33) is synonymous to intermediate precision within Ph. Eur. 5.1.6.
Ph. Eur. 5.1.6 proposes the supplier of the method/technology perform a primary validation using pharmacopoeial test strains appropriate for the intended use and in the absence of product. This would be followed by the end-user verifying some of the supplier’s primary validation data and critical parameters of the method (in the absence of product) using similar pharmacopoeial test strains, in-house isolates or stressed/slow-growing microorganisms. Some of this testing would also include a comparison of the response between the alternative and the compendial methods, which would be analogous to what is described in the USP <1223> equivalency section.
PDA TR33 recommends using a relevant panel of microbial suspensions in a suitable diluent (in the absence of product) to demonstrate the validation parameter acceptance criteria are met, and when applicable, are at least equivalent with the results of the existing method. The latter would also be analogous to what is described in the USP <1223> equivalency section.
Definitions for the validation criteria in Table 1 are provided in the USP <1223> glossary section. However, the chapter particularly discusses specificity, limit of detection, robustness and ruggedness. A summary of USP’s recommendations with a comparison to PDA TR33 and the proposed Ph. Eur. 5.1.6 is provided below.
Specificity
USP <1223> defines specificity as the ability of an alternative method to detect a range or panel of challenge microorganisms specific to the technology. This is similar to the definition provided in PDA TR33 and Ph. Eur. 5.1.6. The panel may be comprised of microorganisms representing a risk to the patient or product, those found in manufacturing environments (e.g., from environmental monitoring), in product (e.g., from in-process bioburden sampling or failed tests) or those that are considered appropriate to utilize for measuring the effectiveness of the method. For growth-based methods, USP recommends to demonstrate the alternative and compendial methods can detect around 100 CFU of each microorganism. This is similar to what Ph. Eur. 5.1.6 recommends for qualitative methods that rely on growth (i.e., to demonstrate presence and absence of microorganisms, “specificity is adequately addressed by demonstrating the growth promotion properties of the media”).
For non-growth-based methods, USP recommends to use negative and positive controls to demonstrate that extraneous matter does not interfere with the ability of the alternative method to detect the panel of microorganisms. This is similarly described in PDA TR33 and Ph. Eur. 5.1.6, although this is particularly addressed under method suitability testing within TR33.
USP also states the challenge microorganisms should be recovered and identified in growth-based methods, and when possible, the same should be done for non-growth-based systems.
PDA TR33 and Ph. Eur. 5.1.6 provide additional guidance that is not specifically mentioned within USP <1223>. For example, TR33 and chapter 5.1.6 both recommend to use mixed cultures and stressed microorganisms, when appropriate and depending on the method and/or application (e.g., using low levels of stressed microorganisms for alternative sterility test methods). TR33 also discusses inclusivity and exclusivity testing for methods that rely on the detection of a specific target microorganism(s) but will not report a positive result if another non-target microorganism is present in the test sample.
Limit of Detection
USP <1223> defines limit of detection (LOD) as the lowest number of microorganisms in a defined volume of sample that can be detected, but not necessarily quantified, under the stated experimental conditions. This is similarly defined in PDA TR33 and Ph. Eur. 5.1.6.
USP <1223> provides two options for demonstrating the LOD. In the first method, microorganisms are diluted to a concentration where the compendial assay will show growth in 50% of the test samples. The diluted samples are then tested in the compendial and alternative methods using a sufficient number of replicates and the following statistically significant risks employed: an alpha risk of 0.05 and a beta risk of 0.20. A Chi-Square test or other appropriate statistical model is used to demonstrate equivalent recovery of the microorganisms. A similar strategy is presented in PDA TR33 whereby the Chi-Square test (when relatively high sample sizes are used), the Fisher’s exact test (when smaller sample sizes are used) or an appropriate statistical test for equivalency is recommended. However, it is unclear why a comparison between the alternative and compendial methods is performed at this time, as USP <1223> states comparisons between the two methods are conducted when demonstrating equivalence (see below) and not necessarily during the qualification of the analytical method itself. The same can also be said for USP’s second LOD option (see below).
The second option for demonstrating LOD employs a Most Probable Number (MPN) strategy where a series of 10-fold dilutions (e.g., from 101 CFU to 10-2 CFU) or 5-fold dilutions (e.g., from 5 CFU to 10-1 CFU) are challenged in the compendial and alternative methods. Five replicates from each dilution are assayed and the MPN is determined from three dilutions in series that provide both positive and negative results (i.e., growth in the compendial method and an appropriate signal in the alternative method). A Chi-Square test or other appropriate statistical model is used to demonstrate equivalent recovery of the microorganisms.
The proposed Ph. Eur. 5.1.6 recommends a similar strategy whereby three 10-fold dilutions are prepared (50 CFU to 0.05 CFU for each challenge microorganism) and ten replicates from each dilution are assayed in the compendial and alternative methods. Using an MPN table, the 95% confidence interval is determined and if the intervals between the two methods overlap, no significant difference for the detection limit of the two methods is observed. The evaluation of LOD should be based on two independent test runs per microorganism and the results of the alternative method should be at least equivalent to those of the compendial method.
PDA TR33 also describes a similar approach where fractional dilutions are utilized. MPN tables are used to calculate the MPN value and upper and lower confidence intervals from not less than 10 replicates per dilution. No significant difference exists between the methods if the confidence levels overlap. TR33 also states if the MPN values permit, a paired t-test or non-inferiority test may be conducted in order to further strengthen the argument of a comparable LOD. This is important to consider especially if the MPN confidence level overlap is very small.
USP defines robustness as the capacity of an alternative method to remain unaffected by small but deliberate variations in method parameters and may be understood as the limits of the operating parameters of the method. An example of deliberate variations in a growth-based method may include departures from normal incubation time and temperature parameters. PDA TR33 and Ph. Eur. 5.1.6 similarly address robustness and all three documents state robustness is a validation parameter that may be addressed through data supplied by the supplier of the alternative method. However, if the end-user modifies a method’s critical parameters, the effects on robustness will need to be evaluated. PDA TR33 and Ph. Eur. 5.1.6 provide additional guidance on how to conduct robustness studies while employing validation criteria appropriate for the type of method under investigation.
Ruggedness
USP <1223> defines ruggedness as the degree of precision of test results obtained by the analysis of the same samples under a variety of typical test conditions such as different analysts, instruments, and reagent lots. PDA TR33 defines ruggedness as the degree of intermediate precision or reproducibility of test results obtained by assessing the same samples under a variety of normal test conditions, such as different analysts, different instruments, different lots of reagents or on different days. According to TR33, intermediate precision is performed within the same laboratory, and reproducibility is performed between laboratories. Ph. Eur. 5.1.6 does not recognize the term ruggedness but instead refers to intermediate precision and reproducibility within its precision sections.
USP <1223> and PDA TR33 state ruggedness is a validation parameter that may be addressed through data supplied by the supplier of the alternative method. However, TR33 provides additional guidance on how to conduct ruggedness testing (as does Ph. Eur. for intermediate precision and reproducibility) while employing validation criteria appropriate for the type of method under investigation.
Demonstrating Equivalency
USP’s new section on equivalency represents the most significant transformation from the previous version of chapter <1223>. Equivalency is intended to show, using standardized microorganism challenges (generally in the absence of actual product), the alternative method is equivalent or non-inferior to the compendial method.
Chapter <1223> states technical support for equivalence may come from peer-reviewed papers or regulatory submissions (e.g., a vendor submitted Drug Master File to the FDA, or prior submission from a company on a technology). However, this may not be sufficient for the manner in which the method will be used and therefore, the end-user may need to determine if additional equivalency testing is required.
The chapter also states statistical evidence is necessary to show equivalence and USP implies a statistical model for demonstrating non-inferiority is appropriate. For example, equivalency may be demonstrated amongst two quantitative methods if there is no statistical difference between the mean cell counts. Yet, USP <1223> claims this may not be possible when the two methods provide different signals, such as a measurement of genomic material or a viability stain fluorescent count, which are not considered a CFU. However, the reader is reminded that other validation guidance documents teach the use of statistical models to compare alternative signals with the CFU when demonstrating equivalency between a novel microbiological method and a classical or compendial growthbased method (see the prior discussion in this article).
USP <1223> provides four different options when demonstrating equivalence. Only three of the options require a direct comparison with the compendial method. Additionally, it is inferred that equivalency is generally demonstrated in the absence of actual product (i.e., product is used during method suitability studies). The four options are as follows:
- The “acceptable procedure” option, in which the alternative method only needs to meet a minimum performance or acceptance requirement and is not compared with the compendial method.
- The “performance equivalence” option, where multiple characteristics of the alternative and compendial methods are compared and an equivalence determination is made based on the results of the comparisons.
- The “results equivalence” option, wherein a single characteristic of the alternative and compendial methods are compared and an equivalence determination is made based on the results of the comparison.
- The “decision equivalence” option, in which a single characteristic of the alternative and compendial methods are compared and an equivalence determination is made based on the conclusions of the analysis.
These four options are intended to offer a variety of opportunities for demonstrating equivalency of an alternative method based on its scientific principle and applications. A closer examination of each option is provided below.
Option 1: Acceptable Procedure
This option does not require a direct comparison between an alternative and a compendial method. Rather, a reference material with known properties is used to demonstrate performance characteristics or acceptance criteria are met. USP <1223> provides examples of reference material, such as a standard inoculum of a specific microorganism, highly purified nucleic acid material, ATP or another “appropriate signal specific to the method.” USP also specifies it may be required to measure the signal in the presence of the test sample using validation criteria that are appropriate for the technology, although there was no explanation under what conditions this would be required. Additionally, USP did not suggest a representative technology or method that would fall into this category of equivalence testing.
Option 2: Performance Equivalence
This option requires a comparison of multiple validation criteria between an alternative and a compendial method. For example, “equivalency or better results” should be shown with respect to validation criteria that are relevant to the alternative method, such as accuracy, precision, specificity, limit of detection, limit of quantification (which appears to be misstated in USP <1223> as ‘limit of qualification’), robustness, and ruggedness. USP also states although an alternative method may not meet certain validation parameters it may still be acceptable for use because of other advantages (e.g., time to result).
This equivalency option appears to be the closest approach to the validation strategies communicated in PDA TR33 and Ph. Eur. 5.1.6 and what many multinational companies have used during the validation of alternative and rapid microbiological methods.
PDA TR33 also addresses when it may be acceptable for an alternative method to be acceptable for use even if a specific validation may not be met. Specifically, TR33 states an alternative method should not be significantly worse than an existing method except when a “clear rationale or justification” exists why the difference can be tolerated.
Option 3: Results Equivalence
This option demonstrates that an alternative and compendial method give an equivalent numerical result. In this option, a tolerance interval is established when comparing the two methods and the alternative method is shown to be numerically superior or non-inferior.
USP discusses the potential for alternative, non-growth-based methods to produce a significantly higher cell count as compared with growth-based methods that produce a CFU. Therefore, when using this equivalency option a calibration curve can be used to show a correlation between the two methods and within an appropriate product or test sample specification range. An example of how this option can be used for an alternative quantitative method is discussed in greater detail in the chapter (see below).
Option 4: Decision Equivalence
This option demonstrates that an alternative and compendial method give an equivalent qualitative result, such as a pass/fail outcome. In this case, the incidence of positive to negative results for an alternative method should be no worse (i.e., the method is non-inferior) to the results obtained with the compendial method. USP also explains this requirement should be based on the historical performance of the material being examined (i.e., a product that has been previously tested and released using the compendial method). An example of how this option can be used for an alternative qualitative method is discussed in greater detail in the chapter (see below).
USP <1223> does not provide guidance when options 3 or 4 (in which a single quantitative or qualitative characteristic is assessed) should be utilized instead of option 2 (whereby multiple characteristics are assessed). One may speculate this would be acceptable for alternative technologies/methods that have been previously and comprehensively validated and described in the literature (or within one’s company) and where a thorough comparison to the compendial method is not required. Nevertheless, further clarification on this point is warranted in a future revision.
Example Approaches for Demonstrating Equivalency
USP <1223> provides example approaches of how to demonstrate an alternative method is equivalent to or better than a compendial method. Although two examples are provided (one for a qualitative method and the other for a quantitative method), USP states that other suitable equivalency strategies may be used as long as they are scientifically justified.
How to Demonstrate Equivalency for an Alternative Qualitative Method
This section of the chapter provides an example of how users can use the fourth option (Decision Equivalence) to demonstrate equivalency for a qualitative method. USP refers to chapters <62> Microbiological Examination of Non-Sterile Products: Tests for Specified Microorganisms, <63> Mycoplasma Tests and <71> Sterility Tests as relevant compendial tests in which a qualitative or a pass/fail result is obtained. Two approaches are provided: a presence/absence test and a Most Probable Number (MPN) test in which qualitative data is converted to quantitative data. Both approaches utilize a one-sided non-inferiority statistical model to demonstrate the alternative method will be as good or better at detecting the presence of microorganisms when compared with the compendial method.
USP explains a non-inferiority approach to equivalence is more appropriate than using a two-sided equivalency model. First, from a patient safety perspective, a non-inferiority approach can encourage the use of an alternative method that is more sensitive than the compendial method. Second, if the alternative method shows better recovery of microorganisms, a two-sided approach may “penalize” the data. Regardless, USP cautions there may be a risk with implementing an alternative method that is more sensitive than the compendial method, especially when the alternative method generates more positive or failing results.
USP also states if an alternative method is not as sensitive as the compendial method but has other advantages, such as a reduced time to result, the alternative method may still be used as long as it “allows for a quality decision on the product that is non-inferior to the compendial method.” However, this strategy needs to be more fully understood especially for critical qualitative assays such as the sterility test. Specifically, if an alternative method does not detect product contamination at a rate at least as good as the compendial test does, can there be a clear rationale or justification for why the alternative method would still be acceptable?
To illustrate this point, the USP microbiology expert committee recently proposed a new rapid sterility test chapter <71.1> in which an alternative method would be capable of detecting product contaminants in the order of magnitude between 10-100 cells (USP Workshop on Alternative Microbiological Methods, March 16-17, 2015, Rockville, MD). Although this range was proposed to encourage certain stakeholders whose products require a rapid sterility test (i.e., compounding pharmacies and the cell therapy industry), it is questionable whether FDA would accept such a proposal considering the historically agreed to limit of detection of a single viable cell (or CFU).
USP’s first qualitative equivalency approach employs the use of presence/absence results. Essentially, a non-inferiority hypothesis is used to demonstrate the proportion of microbial detection in the alternative method is not statistically worse (or inferior) than the proportion of microbial detection in the compendial method. To determine this, the hypothesis uses a specified treatment difference called the non-inferiority margin and this is represented by delta or “Δ.” USP recommends using a Δ = 0.20 (unless there is a reason to use a tighter value) and to calculate a one-sided 90% confidence interval for the microbial detection signal in the alternative method minus the microbial detection signal in the compendial method. Non-inferiority is demonstrated if the lower confidence limit exceeds -Δ or -0.20 at the challenge level under investigation. Additional equations and guidance are provided in the chapter.
Three evaluations are performed using challenge organisms that are representative of what has been recovered from the product, would present a risk to patient and/or compendial organisms. First, serial dilutions are prepared such that a challenge level of 1 CFU is achieved. This will characterize the sensitivity of the alternative method at this concentration. Second, a concentration around 100-200 CFU is used where it is expected that the alternative method will detect the challenge organisms at least 75% of the time. Testing at this level will establish the acceptability of the alternative method. Third, a dilution of organisms around 10-50 CFU is used in both the alternative and the compendial method. The data is used to test non-inferiority as described above. USP recommends using at least 75 replicates for each assessment, providing roughly an 80% test power, or 100 replicates for a 90% test power, if necessary. A greater number of replicates may be required if the alternative method is less sensitive than the compendial method. USP also provides guidance on how to treat the data derived from independent versus paired samples.
The second qualitative equivalency approach employs a standard MPN procedure. Basically, the MPN values obtained by both methods are converted to logs and the sample mean and the sample variance of the log values are determined. USP <1223> provides detailed equations for how to demonstrate non-inferiority when using independent and paired samples.
As a comparison, both PDA TR33 and Ph. Eur. 5.1.6 provide guidance on how to test against specific validation criteria for a qualitative method but instead of employing a single characteristic as is described in USP’s Option 4, both documents generally evaluate all the relevant validation criteria as designated in Table 1 above.
For example, the proposed Ph. Eur. 5.1.6 recognizes the following validation criteria to be considered: specificity, LOD and robustness. Method suitability and equivalency testing with actual product is also identified.
How to Demonstrate Equivalency for an Alternative Quantitative Method
This section of the chapter provides an example of how users can use the third option (Results Equivalence) to demonstrate equivalency for a quantitative method. As described above under Option 3, some alternative methods may provide a different signal than the CFU and this signal may be numerically different in magnitude and units. For example, an alternative method that provides a cell count based on viability staining and laser excitation may provide a “fluorescent count” that is numerically higher than a CFU due to the alternative method being more capable of detecting and enumerating stressed, viable but non-culturable (VBNC) or dormant organisms. In this case, USP recommends demonstrating equivalence between the alternative and the compendial method using two criteria: precision (via repeatability studies) and correlation (via linearity studies).
To demonstrate an alternative method has at least acceptable repeatability, USP recommends using a minimum of six samples with no less than two bioburden levels that are near the specification limit relevant to the application (e.g., if a specification is NMT 100 CFU, use the same concentration for the study). A Chi-Square analysis is performed on the data and repeatability for the alternative method is acceptable as long as the result of the analysis is less or equal to a predetermined specified criterion (i.e., a maximal acceptable repeatability percent geometric coefficient of variation). Because USP does not specify what an acceptable criterion is, it is assumed this would be left to the user to determine.
PDA TR33 states if the coefficient of variation (CV) of an alternative method is less than 35% (this corresponds to a %CV for the traditional plate count method when at least 10 CFU is recovered) there is no need to compare the result to the %CV for a compendial method. However, both TR33 and Ph. Eur. 5.1.6 state regardless of the calculated %CV, the alternative method must have a variance that is not larger than that of the compendial method. If a statistical comparison between %CV’s is required, TR33 recommends to use the McKay approximation (where confidence intervals are compared), a test for equal variance (Bartlett’s test for normal distributed data or Levene’s test for data that is not normally distributed) or a paired t-test.
Next, to demonstrate an alternative method is highly correlated with the numerical results of a compendial method, it is necessary to confirm the acceptance criteria expressed as CFU are calibrated to acceptance criteria expressed as the alternative method’s signal or unit of measurement. To do this, USP recommends to challenge both the alternative and the compendial methods with at least duplicate samples at each of four different bioburden levels covering a range that starts close to the limit of quantification and ends one log higher than a specification limit relevant to the appropriate compendial assay. The correlation (in terms of linearity) is determined by plotting the log values of the recovered microorganisms (e.g., the alternative method on the y-axis and the compendial method on the x-axis). A correlation of at least 0.95 or a R2 value of at least 0.9025 is required to demonstrate linearity between the two methods. In the event the data shows a nonlinear relationship, a Spearman (nonparametric) correlation can be used instead of a Pearson correlation.
However, in the event the required correlation is not met, USP states it may be possible to use Option 3 (Decision Equivalence) as an alternative. In this instance, the user would establish a qualitative acceptance criterion for the alternative method that would match the quantitative specification in the compendial test.
This author previously described a similar strategy (called the “dilute-to-spec” method) for using a qualitative method to predict meeting a quantitative specification.11 For example, if a compendial specification is not more than 1000 CFU, it may be possible to correlate a qualitative result to match this required level of microbiological quality by diluting a test sample to this same specification level (i.e., diluting 1:1000). If the qualitative result is negative (no growth or microbial recovery), the user has demonstrated that the number of microorganisms in the original test sample was less than 1000, and the compendial specification has been met. However, if the number of microorganisms in the original sample is very close to the specification level, this strategy may not be appropriate to use because the qualitative result can provide either a positive or a negative result, depending on the actual distribution of microorganisms or other factors. Therefore, an understanding of the historical bioburden level in the test sample may help to resolve this potential issue; otherwise, confirmatory testing may be required. This is exactly the approach GSK previously utilized with a qualitative ATP bioluminescence technology to release a non-sterile nasal spray that had a quantitative Microbial Limits specification: if a positive qualitative result was obtained, the required specification was confirmed using the quantitative, compendial assay. GSK also used the same strategy for testing biological indicators.12
PDA TR33 and Ph. Eur. 5.1.6 provide guidance on how to test against specific validation criteria for a quantitative method but instead of employing a single characteristic as is described in the USP’s Option 3, both documents generally evaluate all the relevant validation criteria as designated in Table 1 above.
For example, the proposed Ph. Eur. 5.1.6 specifies the following validation criteria for a quantitative method: accuracy, precision, specificity, limit of quantification (LOQ), linearity, range and robustness. Method suitability and equivalency testing with actual product is also required.
Method Suitability
In this article, we have reviewed USP’s recommended phases of alternative method validation: qualification of the instrumentation, qualification of the analytical method and a demonstration of equivalence (between the alternative and the compendial method). The latter two phases generally utilize standardized microbial suspensions in the absence of product or test samples.
The user must now determine whether actual product or test samples that will be introduced into the validated alternative method will not inhibit or enhance an alternative signal with respect to certain validation criteria. USP <1223> describes these activities as Method Suitability.
Specifically, the chapter states for each new product to be evaluated with a validated alternative method, suitability testing as prescribed in USP <51>, <61>, <62>, <63> and <71> should be performed using the same sample preparation, quantity and number of units appropriate for the product and the required level of assay sensitivity. This testing should demonstrate the alternative signal (i.e., relating to microbial detection or quantification) is not quenched or increased in the presence of the product being evaluated. PDA TR33 and Ph. Eur. 5.1.6 also describe the use of actual product or test samples for this purpose. However, there may be some confusion over the terminologies being used in each document.
For example, while USP identifies actual product to be used to demonstrate method suitability, TR33 uses actual product during method suitability and equivalency testing:
Method Suitability (TR33): “To demonstrate that the new method is compatible with specific product or sample matrices that will be routinely assayed, each material should be evaluated for the potential to produce interfering or abnormal results, such as false positives (e.g., a positive result when no viable microorganisms are present in the test sample) or false negatives (e.g., a negative result when microorganisms are present in the test sample). This may also include evaluating whether cellular debris, dead microorganisms or mammalian cell cultures have any impact on the ability of the new method and accompanying system to operate as it is intended to.”
Equivalence/Comparative Testing (TR33): “Equivalence or comparative testing involves the use of actual product and other sample matrices that will be routinely tested using the alternative or rapid method once it is validated and implemented.”
Similarly, the proposed Ph. Eur. 5.1.6 uses actual product during suitability testing and the demonstration of equivalence:
Suitability Testing (5.1.6): “The alternative method must be applied according to the specified procedure and with the samples to be analysed under the responsibility of the user. The method must be shown to give comparable results as characterised in the model system used by the supplier. Compatibility of the response with the product prepared as needed by the user, evaluated using pharmacopoeial test strains.”
Equivalence Testing (5.1.6): “A direct approach to demonstrating the equivalence of two qualitative methods would be to run them side-by-side and determine the degree to which the method under evaluation leads to the same pass/fail result as the pharmacopoeial method. This parallel testing shall be performed based on a prespecified period of time or number of samples.”
Due to the differences in when and how product is used within each validation guidance document, it is necessary to briefly explore how each document refers to method suitability and equivalence.
A Comparison of Method Suitability Strategies
USP <1223> recommends three (3) independent tests to be conducted to demonstrate method suitability. Only accuracy and precision are required to be evaluated for quantitative methods and the recovery of microorganisms according USP ,62>, <71> and <1227> is all that is required for qualitative methods. Furthermore, once an alternative PDA TR33 similarly states it is the end-user’s responsibility to ensure that all test samples will be compatible with the new or alternative method. However, neither TR33 nor Ph. Eur. 5.1.6 specifically restricts the assessment of the relevant validation criteria to a single product. Separately, TR33 describes a reduced validation strategy when transferring a validated, like-for-like alternative method to a secondary testing site. This may include testing a few reference organisms, identical to what was used during the original qualification, as well as some local facility isolates to confirm basic functionality and demonstrate that key qualification requirements are met (e.g., accuracy and precision). Nevertheless, even though the original equivalency testing may assess the same type of test sample as what the secondary site will be evaluating routinely, the microbial load (number and type of microorganisms) may not be the same. Therefore, equivalency testing may need to be conducted using the actual test material at the secondary site.
When speaking of the potential for false positive and false negative results, PDA TR33 explains a product or test sample could cause an improper indication of the presence of microorganisms if it contains material that produces background noise or interfering signals. Furthermore, a product or sample matrix containing material that may quench, mask or otherwise prevent the detection or enumeration of microorganisms when they are present could cause an improper indication that microorganisms are absent. When testing for false positives, sterile test samples should be utilized, when possible. When testing for false negatives, samples with a known quantity and type of microorganisms should be used. Although no specific criteria to be evaluated are identified during these studies, similar strategies as recommended by USP may be employed (e.g., evaluating accuracy and precision for quantitative methods; LOD or inclusivity/exclusivity for qualitative methods).
For suitability testing, the proposed revision to Ph. Eur. 5.1.6 recommends assessing the detection limit for qualitative methods and accuracy, LOQ and linearity for quantitative methods. Acceptance criteria for the method in routine use will need to be defined as a function of the application and the validation data. More specifically, chapter 5.1.6 provides an example of how to conduct suitability testing for a qualitative method (i.e., a rapid ATP bioluminescence sterility test) and a quantitative method (i.e., solid phase cytometry using membrane filtration, fluorescent cell labeling and laser scanning):
Suitability of a qualitative method is demonstrated using an adequate rinse protocol to confirm the product of interest does not interfere with the alternative method by inducing a high bioluminescent background (false positive) or inhibiting the bioluminescence reaction (false negative). Inhibition of the bioluminescence reaction can be excluded by inoculating 3 membranes through which the drug product of interest had been filtered with 10-100 CFU of a stressed, slow-growing microorganism and the recovery of a predefined number of bioluminescent microcolonies is similar as the control (identically inoculated membranes through which no product had been filtered).
Suitability of a quantitative method is demonstrated by separately adding not more than 100 CFU for each test microorganism into the rinse (at the same rinsing step and in the same manner as used for the compendial method) of the membrane filtration step for both the alternative and compendial methods. A recovery of 50-200% in accordance with Ph. Eur. 2.6.12, Microbiological Examination of Non- Sterile Products: Microbial Enumeration Tests, is required.
A Comparison of Equivalence Testing Strategies
USP <1223> recommends performing equivalency testing using standardized microorganism challenges and applicable validation criteria, depending on the equivalency option utilized. The chapter infers this testing is generally performed in the absence of actual product or test samples. Conversely, PDA TR33 and Ph. Eur. 5.1.6 use actual product or test samples during the demonstration of equivalency.
TR33 specifies product or test samples are assessed in parallel for a specified period of time or number of product batches. Similar procedures and data analyses previously utilized for the validation criteria with standardized cultures in a suitable diluent can be used (e.g., accuracy, precision, LOQ, LOD, linearity or range). To best illustrate this strategy, Novartis demonstrated equivalence between a rapid ATP bioluminescence sterility test and the compendial growth-based method by conducting 90 sterility tests in both methods in which one drug product was inoculated with 1-5 CFU of three different stressed microorganism strains.13 The Chi Square test, Fisher’s exact test and the Fisher test for a one sided equivalence was utilized during the statistical analyses.
For equivalence testing, the proposed revision to Ph. Eur. 5.1.6 states to run the alternative and the compendial methods side-by-side on a predefined period of time or number of samples. For a qualitative method, the degree to which the method under evaluation leads to the same pass/fail result as the compendial method is determined. However, this approach may not be suitable for an alternative sterility test, because the number of positive samples will be extremely low. In this instance, samples of representative drug products are inoculated with a very low number (e.g., less than 5 CFU) of at least three different strains of test microorganisms. For each test method, product and test microorganism, a minimum of three test runs with a sufficient number of replicates are performed. A comparison of the pass/fail results shows the alternative method to be at least equivalent to the pharmacopoeial method.
However, for a quantitative method, if the result of the alternative method can be expressed as a number of CFU per weight or per volume, statistical analysis of the results shall demonstrate that the results of the alternative method are at least equivalent to those of the compendial method. If the result of the alternative method cannot be expressed as a number of CFU, then statistical analysis shall demonstrate that the results of the alternative method are at least equivalent to those of the compendial method.
The proposed Ph. Eur. 5.1.6 provides an example of how to conduct equivalence testing for an ATP-based qualitative sterility test and a quantitative solid phase cytometry test:
Equivalency of a qualitative test is demonstrated by artificially contaminating sterility test samples with less than 5 CFU of three different test microorganisms. Samples are treated using the same rinse protocols and examined side-by-side in both methods. For each test method (product and microorganism), three test runs with 10 replicates each are performed. The results are statistically evaluated to demonstrate the alternative method is at least equivalent to the compendial method.
Equivalency of a quantitative test is demonstrated by performing the alternative and compendial methods side-by-side. The enumerative results (CFU and cell counts) are statistically evaluated to demonstrate the alternative method is at least equivalent to the compendial method.
Based on this comparative review, it appears that the USP’s use of actual product during the demonstration of method suitability is included in both method suitability and equivalence within PDA TR33 and Ph. Eur. 5.1.6. Additionally, USP’s use of standardized microorganisms during the demonstration of equivalency is incorporated in validation criteria testing and the verification of primary validation as described in PDA TR33 and Ph. Eur. 5.1.6, respectively.
Summary
The 2015 revision to USP <1223> affords a significant departure from the original version published in 2006. Some of the changes mirror the teachings in PDA TR33 and the proposed Ph. Eur. 5.1.6 while others provide a substantial shift in the way alternative methods might be validated for their intended use. Therefore, end-users should weigh the similarities and differences when formulating a meaningful and defendable validation strategy and most importantly, appreciate the opportunities for implementing alternative and rapid microbiological methods.
Acknowledgment
The author thanks Jeanne Moldenhauer, Excellent Pharma Consulting, for her exceptional review of this manuscript.
References
- USP. 2015. <797> Pharmaceutical Compounding – Sterile Preparations. United States Pharmacopeial Convention. USP 38/NF33:567.
- PDA. 2000. Evaluation, Validation and Implementation of New Microbiological Testing Methods, Technical Report No. 33, Parenteral Drug Association, Vol. 54, No. 3, May/June 2000, Supplement TR33
- PDA. 2013. Evaluation, Validation and Implementation of Alternative and Rapid Microbiological Methods, Technical Report No. 33 (Revised 2013), Parenteral Drug Association
- Ph. Eur. 2006. Chapter 5.1.6 Alternative Methods for Control of Microbiological Quality, European Pharmacopeia, European Directorate for the Quality of Medicines & HealthCare. 5.5:4131.
- Pharmeuropa. 2015. Chapter 5.1.6 Alternative Methods for Control of Microbiological Quality, European Directorate for the Quality of Medicines & HealthCare. 27.1:8
- Wicks, S.J. 2015. The Role of the European Pharmacopoeia in the use of Alternative Microbiological Methods. Alternative Microbiological Methods: A Workshop on Current Status and Future Directions of Compendial Standards. March 16-17, 2015. Rockville, MD.
- USP. 2006. <1223> Validation of Alternative Microbiological Methods. United States Pharmacopeial Convention. USP 29/NF24, Suppl. 2:3807.
- USP. 2014. <1223> Validation of Alternative Microbiological Methods. United States Pharmacopeial Convention. USP 37/NF32:1152.
- USP. 2015. <1223> Validation of Alternative Microbiological Methods. United States Pharmacopeial Convention. USP 38/NF33:1439.
- USP. 2015. <1058> Analytical Instrument Qualification. United States Pharmacopeial Convention. USP 38/NF33:971.
- Miller, M.J. 2012. Case Study of a New Growth-Based Rapid Microbiological Method (RMM) that Detects the Presence of Specific Organisms and Provides an Estimation of Viable Cell Count. American Pharmaceutical Review. 15(2): 18-25.
- Dalmaso, G. 2006. Rapid Steam Sterilization Biovalidation Using Biological Indicators and the Pallchek Luminometer. Encyclopedia of Rapid Microbiological Methods Volume II, edited by Michael J. Miller, PDA/DHI Books. 251-272.
- Gray, J.C.; Staerk, A.; Berchtold, M.; Mercier, M.; Neuhaus, G.; Wirth, A. 2010. Introduction of a Rapid Microbiological Method as an Alternative to the Pharmacopoeial Method for the Sterility Test. American Pharmaceutical Review. 13(6): 88-94.
Author Biography
Dr. Michael J. Miller is an internationally recognized microbiologist and subject matter expert in pharmaceutical microbiology and the design, validation and implementation of rapid microbiological methods (RMM). He is currently the President of Microbiology Consultants, LLC (http://microbiologyconsultants.com) and owner of http://rapidmicromethods.com, a website dedicated to the advancement of rapid methods.
For more than 25 years, he has held numerous R&D, manufacturing, quality, business development and executive leadership roles at multinational firms such as Johnson & Johnson, Eli Lilly and Company and Bausch & Lomb. In his current role, Dr. Miller consults with multinational companies in providing technical, quality, regulatory and training solutions in support of RMMs, sterile and non-sterile pharmaceutical manufacturing, contamination control, isolator technology, environmental monitoring, sterilization and antimicrobial effectiveness.
Dr. Miller has authored more than 100 technical publications and presentations and is the. He currently serves on the editorial and scientific review boards for American Pharmaceutical Review, European Pharmaceutical Review and the PDA Journal of Science and Technology. Dr. Miller holds a Ph.D. in Microbiology and Biochemistry from Georgia State University (GSU), a B.A. in Anthropology and Sociology from Hobart College, and is currently an adjunct professor at GSU.