Analytical Sciences, Pharmaceutical Sciences and Clinical Supply
Analytical Sciences, Pharmaceutical Sciences and Clinical Supply
Analytical Development and Commercialization
Drug products for pediatric patients are typically formulated as powder for suspension, oral granules, oral solutions and most recently mini-tablets. In the past, most pharmaceutical companies deferred pediatric formulation development long after the adult product had been marketed, with this work mainly driven by receiving additional market exclusivity in major markets. However, agency expectations make it mandatory for pharmaceutical companies to communicate a pediatric formulation assessment for new pharmaceutical products, which often leads to pediatric formulation development activities even prior to application of the adult formulation1. Developing a pediatric drug product is often challenging since a single product may not be feasible for all pediatric age groups including neonates, infants, toddlers, young children, and adolescents. Mini-tablets offer an elegant single formulation approach to address the unique dosing considerations of each of these pediatric patient groups. For the caregiver and ultimately the patient, the advantages of mini-tablets are generally ease of use, simple dose adjusting provided by packaging, and accurate dose delivery.
The development of a mini-tablet formulation can be very challenging especially if the regulatory filing strategy for the pediatric formulations include demonstration of bio-equivalency to the adult formulation. Since the pediatric formulation is often developed after the adult formulation process is optimized, pharmaceutical companies are often compelled to use similar formulation and process intermediates which may not be optimized for a mini-tablet formulation. In order to ensure the mini-tablet process is robust, the formulation development team typically faces two main issues: ensuring acceptable tablet content uniformity and dissolution performance. These attributes are mainly impacted by the tooling design, which is similar to conventional tablet compression tooling but provides a much larger number of tablets per compression event, which are often the size of typical granules. Obviously, analyzing individual mini-tablets for content uniformity and dissolution provides the most informative data to guide process development. As there may be slight differences in fill levels amongst die openings, using multi-tip tooling can cause variation within each compression event. Considering both this extra source of variability and the large number of units per batch, the number of units required for testing to provide statistically meaningful data can quickly become overwhelming compared to traditional tablet development. To address the larger sample volume with mini-tab development, we employed auto-mation to assess content uniformity and rate of dissolution. Furthermore, we approached dissolution method develop-ment in a Design of Experiment (DOE) approach, using automation to complete this work in a comp-rehensive and efficient manner.
Advantages of Mini-Tablets
In a clinical study, one mini-tablet formulation can easily deliver the same weight adjusted dose, on an mg per kg basis, to all patients by simply counting out the prescribed number of mini-tablets for each patient at the clinical site. Although accurate administration of a liquid formulation is easily accomplished in a clinical setting, this is often a concern with actual patient use. Studies have shown that significant training or oversight from the pharmacist or doctor, is often necessary to ensure that a patient or one word can accurately deliver the intended dose using a syringe2. Use of mini-tablets makes accurate dosing far easier for the patient. Mini-tablets are packaged into a stick pack or sachet and are delivered by opening the package and dumping the entire contents of the appropriate number of stick packs onto a soft food for patient consumption. This level of patient convenience is a significant advantage for a mini-tablet formulation.
 Figure 1. Examples of multi-tip tooling typically employed in the manufacturing of mini-tablets. At left is a set of upper punches with corresponding dies for 2, 3 and4 tip tools; in the middle, a 12-tip punch; at the right is a complete set of punches and die for a 7-tip tool. As with shaped tooling, the multi-tip mini-tablet tooling is keyed to maintain mini-tablet rotational orientation.
Figure 1. Examples of multi-tip tooling typically employed in the manufacturing of mini-tablets. At left is a set of upper punches with corresponding dies for 2, 3 and4 tip tools; in the middle, a 12-tip punch; at the right is a complete set of punches and die for a 7-tip tool. As with shaped tooling, the multi-tip mini-tablet tooling is keyed to maintain mini-tablet rotational orientation.Patient acceptability and palatability is a significant concern with pediatric patients. Many Active Pharmaceutical Ingredients (API) have a bitter or otherwise unpleasant taste. Liquid, suspension, or other flavored formulations may block the taste of the API with another strong flavor, which may still be rejected by some patients. Instead of adding a flavoring, blocking the taste of the API enables a flavorless formulation. This is a preferred option as it allows the patient or care-giver to simply add the mini-tablets to their food of choice, which should increase patient acceptability levels. In a study aimed to evaluate the acceptability of mini-tablets, patient acceptance of 2-mm mini-tablets was at least equal to or even better than that of sweet tasting syrup3.
Impact of Multi-Tip Tooling on Development
Due to the low amount of API per mini-tablet, using single-tipped tooling for compression is not practical as the run times, even for a small volume product, would be exceedingly long. For example, if a tablet press was set up with 50 tools and operated at 20 RPM, this would produce 60,000 tablets in one hour. Assuming a pediatric unit dose requires packaging 50 mini-tablets in one stick pack, this would equate to 1200 stick packs per hour. Assuming 30 doses are packaged for a 30 day supply of the product to one patient, this means the manufacturing process is producing enough product per hour to supply 40 patients with a 1 month supply. Use of multi-tip tooling can greatly increase productivity and reduce this run time. Following the example above, use of tooling with 10 tips per punch instead of only 1, would reduce the time needed by an order of magnitude. While the use of multi-tip tooling greatly improves throughput, it presents additional challenges for development.
When using multi-tip tooling, multiple mini-tablets are made per compression event. As such, the consistency with which each die opening is filled will have an impact on the uniformity of both the weight and the hardness. A consistent powder or granule flow will greatly help to ensure adequate uniformity. As some variation in fill and therefore hardness is inevitable, a robust formulation should exhibit minimal changes in dissolution across a reasonable range of compaction force. As multi-tip tooling incurs this additional source of potential variability, additional mini-tablets should be tested, per batch, in order to ensure a statistically meaningful result with a reasonable confidence level. In addition to typical attributes to be assessed, different mini-tablet tooling designs may also be evaluated during formulation development. Given these considerations, sample size can quickly become unmanageable with a traditional “one-tabletper flask” testing approach even if a large number of analytical staffcan be committed to support testing. Automation can greatly enhance productivity, analyzing samples overnight and through the weekends, enabling a sufficient quantity of data to be obtained with minimal oversight by laboratory personnel.
Challenges with Content Uniformity for Mini-Tablet Development
Although mini-tablets present some clear advantages for the patient, development of a mini-tablet formulation poses unique challenges with respect to consistent dose assay and uniformity. As each patient will receive tens of mini-tablets per dose, the number of units produced per batch is much larger than typical for each stage of development. As mini-tablets are designed to be sprinkled upon a soft food for consumption, they should meet the FDA’s guidance for a sprinkle formulation of no larger than 2.5 mm4. As it can be difficult to deliver a formulation and a process which can accurately and consistently fill a die opening of no more than 2.5 mm; content uniformity will be a significant concern through-out development.
An example 2-mm mini-tablet, shown in fig-ure 2, has an average mass of approximately 6.5 mg. Considering this small image, a 1 kg batch size, reasonable for early development, would therefore have a theoretical yield of over 150,000 units. Typically for traditional tablet sizes, batches of this number of units would only be produced far later in the development space. In order to characterize a batch of this size, a larger number of tablets should be analyzed. Given the small size of the mini-tablets, very small changes to tablet mass will cause significant changes to the assay. In total, these differences place a larger emphasis on content uniformity testing than for a larger tablet image.
During early development, the appropriate number of mini-tablets to be tested per sample preparation must be considered. For a marketed product, it is appropriate to treat one packaged unit, such as a sachet or stick pack, as one unit since this represents one patient dose. It follows this logic that for early clinical release testing, the lowest planned clinical dose, perhaps 5-10 mini-tablets, should be treated as one unit for analysis. However, we suggest to best guide process development and optimization, these initial analyses should be performed on individual units. Performing content uniformity testing of individual mini-tablets will provide a greater level of discrimination to the various formulation options, but unfortunately this approach will greatly increase the number of analyses required.
 Figure 2. 2-mm mini-tablets shown with a US penny and a metric ruler for size comparison.
Figure 2. 2-mm mini-tablets shown with a US penny and a metric ruler for size comparison.Automation presents an attractive option to handle large sample volumes with minimal oversight and effort from laboratory per-sonnel. For content uniformity or assay testing, the analyst needs only to load the samples into the instrument and pre-pare a sufficient volume of diluent. The auto-mation systems will obtain the sample mass, quantitatively deliver the appropriate volume of diluent and then extract the sample through use of a homogenizer or blender. After mixing, the sample is filtered, diluted further if necessary, and then either vialed or directly injected onto an HPLC. Programming these automated systems to run overnight or through the weekend can allow for a small number of analysts to complete testing of this larger number of required samples reliably and within a reasonable time frame.
Dissolution Challenges
As with other dosage forms, dissolution testing for mini-tablets can be critical for screening prototype formulations and, if required, guide formulation development with the goal to meet bio-equivalence to the adult formulation. Development of a robust dissolution method which would be useful for the characterization of mini-tabs and potentially serve as a quality control method can be a complex exercise that must examine multiple aspects, including solubility of the active pharmaceutical ingredient (API), solubility of a film coating agent (if employed), formulation composition, and compression force of the dosage form itself. Similar to larger dosage forms, dissolution method development must consider factors such as: the pH of the media employed, buffer strength of the media, and level of surfactant used (if any). As with content uniformity testing, automation presents an attractive option for dissolution testing of a large number of samples and for the rapid development of a method where the experimental conditions need to be justified in a multi-variate (DOE) approach.
 Figure 3. Example DOE used to inform dissolution media selection.
Figure 3. Example DOE used to inform dissolution media selection.An example Design of Experiments approach (DOE) for the development of a dissolution method is shown in Figure 3 for media selection with a formulation which employed a film coating for taste masking. In this case study, the formulation also has a pH-dependent solubility, with a faster dissolution in neutral pH than in acidic media. This DOE was executed in an automated fashion to rapidly characterize each variable to ultimately arrive at a finalized set of method conditions4. In order to execute this DOE, the analyst needed only to prepare the various media and load the mini-tablets into the appropriate sample holder. The automated dissolution system fills the vessels, heats the media to 37˚C, drops the mini-tablets, and then pulls and filters samples at each time point into HPLC vials or directly injects them onto an HPLC. At the conclusion of the run, the automated system empties and cleans the vessels prior to running the next dissolution test. Certain automated dissolution systems can accommodate and switch amongst four or more different media, selecting the appropriate media per the program set by the analyst. The entire cycle time of filling, heating and cleaning takes approximately one hour; so each one-hour dissolution requires about 2 hours of instrument time, but very little analyst time6.
By utilizing this DOE approach, a suitably discriminating method was generated very quickly and allowed the streamlined support of process development. The DOE shown in Figure 3, a half-factorial design with added points at likely media of interest, requires 102 dissolution experiments, generating N=3 each for coated and uncoated tablets in each media. Execution of this in a short time period is a challenge if done manually; however, by using automation, the experiments can be run overnight or over the weekend without analyst touch time, greatly reducing methods development time. This dissolution method is adequate to guide formulation and process screening/development and the comprehensive data set provided a solid base for any further method optimization once the final formulation and process is further developed.
Dissolution in this selection of media was performed for both uncoated and film-coated tablets with an analysis of the important factors done using a commercially available statistical software program. An analysis of the responses from the DOE shows that factors which are statistically significant can vary from uncoated to coated tablets. Figures 4 and 5 show that pH is clearly the most significant factor on the dissolution for both coated and uncoated mini-tablets in this formulation. For the uncoated mini-tablets, the surfactant level also has a significant impact on the dissolution rate. However, for the uncoated mini-tablets, the ionic strength of the media has a minor impact and the surfactant level is no longer significant. Performing a DOE in this manner can provide the development team with a wealth of information as to what impacts the dissolution of the formulation. Use of automation enabled this work to be performed by a single analyst in approximately one week.
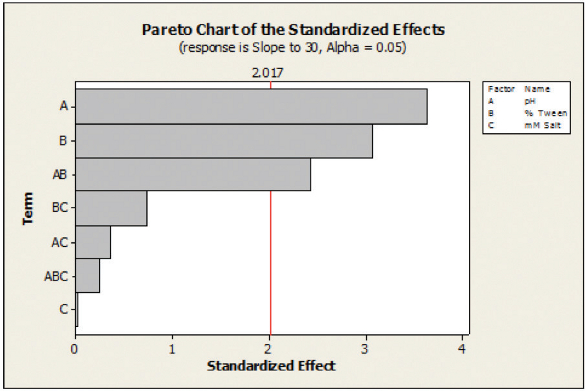 Figure 4. Pareto Chart showing the standardized eff ects, with response of dissolution release to 30 minutes, for uncoated tablets.
Figure 4. Pareto Chart showing the standardized eff ects, with response of dissolution release to 30 minutes, for uncoated tablets.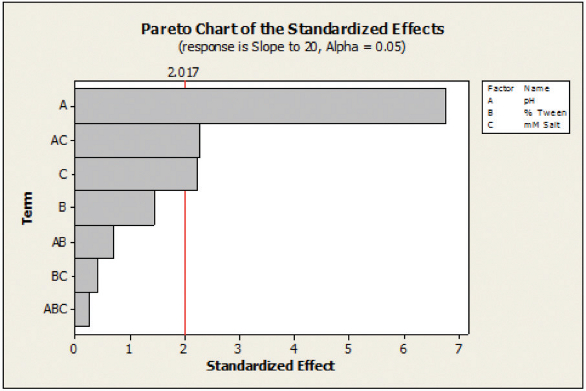 Figure 5. Pareto Chart showing the standardized eff ects, with response of dissolution release to 20 minutes, for coated tablets..
Figure 5. Pareto Chart showing the standardized eff ects, with response of dissolution release to 20 minutes, for coated tablets..Conclusion
Development of a mini-tablet formulation involves several unique challenges which may require extensive content uniformity and dissolution testing. As the number of samples tested in the development space expands, one may seek to employ automated technologies to aid in the rapid and efficient testing of content uniformity and dissolution samples. Out-of-the-box technologies, such as automated extraction platforms for preparation of content uniformity samples as well as dissolution testers that fully automate the dissolution test, are readily available on the market and can be implemented easily in most settings. Used in conjunction, such technologies can aid in generation of vast amounts of data that can greatly expedite the development process. As development of a minitablet formulation may require testing of a larger number of samples than for a larger dosage form, incorporating automated technologies becomes even more valuable for formulation development.
References
- U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER) July 2013, “Guidance for Industry Pediatric Study Plans: Content of and Process for Submitting Initial Pediatric Study Plans and Amended Pediatric Study Plans.”
- McMahon, Shawn R., Rimsza, Mary E., Curtis Bay, R. “Parents Can Dose Liquid Medication Accurately” Pediatrics January January 29, 1997
- Sporner, Natalie, Breitkreutz, Joerg, et al. “Acceptance of uncoated mini-tablets in young children: results from a prospective exploratory cross-over study.” Arch Dis Child 2012, 97:283-286
- U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER) May 2012 CMC Rev. 1 “Guidance for Industry Size of Beads in Drug Products Labeled for Sprinkle.”
- Brian Kozlowski “Automated Dissolution Testing” Encyclopedia chapter in the Encyclopedia of Pharmaceutical Science and Technology, 4th Edition
- Brian Kozlowski and Pete Wuelfing. “Automation of Dissolution – An Evaluation of Existing Technologies and Approach to Implementation.” American Pharmaceutical Review, May/ June 2007.
Author Biographies
Francis Flanagan is an Associate Priniciple Scientist at Merck & Co., Inc. with over 15 years experience in research within the pharmaceutical industry. His primary responsibilities include leading anayltical efforts in support of the development of new pharmaceutcial compounds. Throughout his career at Merck, Fran has supported a variety of programs including Vytorin and Belsomra. His reseach interests include furthering our understanding the impact of hot melt extrusion on pharmaceutical product quality. Fran holds a B.S in Chemistry from Loyola University in Maryland.
Brian Kozlowski is a Associate Principle Scientist at Merck Sharp and Dohme Corp. with fifteen years experience in the pharmaceutical industry. Brian is responsible for the development and validation of analytical methods used in stability characterization and release testing of finished pharmaceutical dosage forms. Additionally, Brian is a member of an automation technology group in the Analytical Sciences Department, spending a large portion of his efforts on evaluation and implementation of automation for analysis of tablets and capsules. Brian holds a B.S. degree in Chemistry from Mount St. Mary’s University in Emmitsburg, MD, and an M.S. in Pharmaceutical Chemistry from Lehigh University in Bethlehem, PA.
Erin Heinis a Scientist at Merck & Co. with 9 years experience in the pharmaceutical industry. Erin is responsible for the development and validation of analytical methods used in stability characterization and release testing of finished pharmaceutical dosage forms. Additionally, Erin is a member of an automation technology group in the Analytical Sciences Department, spending a large portion of her efforts on evaluation and implementation of automation for analysis of tablets and capsules. Erin holds a B.S. degree in Chemistry from Temple University in Philadelphia, PA.
Gary Romberger is a Scientist at Merck Sharp and Dohme Corp. with eleven years experience in the pharmaceutical industry. Gary is responsible for the development and validation of analytical methods used in stability characterization and release testing of finished pharmaceutical dosage forms. Additionally, Gary is a member of an automation technology group in the Analytical Development in Commercialization, spending a large portion of his efforts on evaluation and implementation of automation for analysis of tablets and capsules. Gary holds a B.S. degree in Chemistry from Lebanon Valley College in Annville, PA.