Note - this article is an overview of a topic originally presented at the 2015 American Association of Pharmaceutical Scientists Annual Meeting in Orlando, Florida.
Introduction
Contemporary small molecule therapeutics are often hindered by poor physicochemical properties1,2, with high lipophilicity and/or crystallinity leading to poor aqueous solubility. This in turn limits oral absorption resulting in low or variable systemic exposure. While the concept of poor solubility is well understood, a practical definition is more complex. The USP-NF defines several categories of solubility limits. For instance, the lowest category deems a compound to be practically insoluble/insoluble if the solubility is less than 0.1mg/mL in water3. Alternatively the biopharmaceutical classification system categorizes solubility relative to dose and also includes dissolution rate criteria, such that a drug is considered soluble if4:
i) the highest dose dissolves into 250mL of aqueous solution across the physiological pH range of 1 to 7.5, and
ii) dissolution in 900mL is 85% complete within 30 minutes
Although specific, definitions such as those above are of limited utility for optimizing and selecting drug candidates, since the solubility of many discovery phase compounds are often lower than 1 μg/mL and clinical doses have not been determined. Furthermore, such static measurements do not fully account for the variable conditions within the gastrointestinal tract - solubility in complex and variable gastric or intestinal fluids, the gastric to intestinal pH shift, dilution of solution formulations as well as the potential for supersaturation and precipitation.
Fortunately, applying solubility enabling formulation strategies can help ensure drug is sufficiently dissolved within the intestinal environment for adequate drug absorption. Relevant influencing factors and several in-vivo case studies are discussed further below.
Discussion
The Gastrointestinal Environment
Within the gastrointestinal environment there are multiple factors influencing drug concentrations. For instance, as a drug transitions along the gastrointestinal tract differences in volumes, motility rates and fluid compositions (ie. bile, pH, food) can lead to significant differences in solubility and concentration between test subjects5,6. Such variability should be taken into account when developing and assessing potential formulations.
Formulation performance is typically evaluated with in-vitro modeling systems. The degree of sophistication for the model depends on the phase of drug discovery or development. In the early stages of discovery, the number of compounds to evaluate and limited material available necessitates simple, efficient experiments. For instance, testing solubility and precipitation potential in simulated biorelevant fluids such as FaSSIF7 by turbiditimetric or supernatant analyses8,9. Later stages of drug candidate selection and advancement warrant more sophisticated gastrointestinal modeling such as modified dissolution systems10,11 which may also incorporate a permeable membrane to simulate absorption12. Multi-compartment dissolution/absorption models such as the TNO TIM system13 may also be brought to bear as a drug candidate approaches clinical trials.
The Free Drug Hypothesis
While a drug must be in solution to be absorbed, the free drug hypothesis postulates that only the free molecular species can permeate across the intestinal membrane14. In the case of a simple suspension, the free drug concentration is typically low (Figure 1, center). For a solubilized formulation, more total drug is present in solution (Figure 1, right). However most of the drug is bound within the solubilizer matrix (ie. a cyclodextrin cavity or a surfactant micelle) and is therefore unavailable for absorption directly. Instead the solubilized portion of the dose acts as a replenishment reservoir to rapidly resupply the free drug depleted by absorption. Alternatively supersaturating formulations increase both the total and free drug concentrations, enabling greater absorption for as long as the supersaturated state persists (Figure 1, left).
 Figure 1. Schematic Illustrating Total Versus Free Drug Concentrations in Suspension, Solubilization and Supersaturation Systems
Figure 1. Schematic Illustrating Total Versus Free Drug Concentrations in Suspension, Solubilization and Supersaturation SystemsFree drug concentrations are significantly reduced when an excessive amount of solubilizer is used relative to the amount of drug and can reduce drug absorption. This has been demonstrated in a series of in-vitro experiments comparing solubilized and supersaturated systems15-17. These studies provide an excellent example of the risk of oversolubilization, albeit under sink conditions at concentrations lower than those resulting from typical in-vivo doses. In general, a crystalline suspension often represents a worst case against which to compare solubility enabling formulations. Oral absorption and exposure may be improved even with imperfect non-optimized solubilizing or supersaturating delivery systems.
Solubility Enablement Strategies - Solubilization vs. Supersaturation
Solubility enablement is often necessary for compounds demonstrating solubility limited oral absorption. This encompasses two distinct but complimentary strategies - solubilization and supersaturation. These two approaches are distinguished by the impact of aqueous dilution upon the concentration, relative to the equilibrium solubility within the diluted mixture. Both the concentration and solubility of a solubilized formulation dilute linearly, so the concentration never exceeds the solubility (Figure 2a). The concentration of a supersaturated formulation also dilutes linearly, however the solubility decreases exponentially with dilution (Figure 2b). This creates a kinetically unstable state in which the concentration exceeds the equilibrium solubility. This unstable state provides a driving force for the compound to leave the solution - beneficial for oral absorption but potentially detrimental when the result is precipitation.
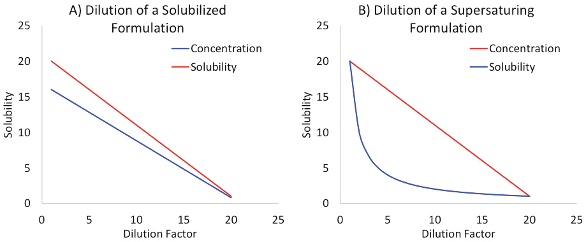 Figure 2. The Effect of Aqueous Dilution on Concentration and Solubility for Solubilized vs Supersaturating Systems
Figure 2. The Effect of Aqueous Dilution on Concentration and Solubility for Solubilized vs Supersaturating SystemsSolubilized solutions overcome dissolution limited solubility (Figure 3a) by predissolving the compound in delivery vehicles containing modified cyclodextrins, surfactants or non-digestible emulsifiers. The total drug in solution is increased (ie. often the entire dose is predissolved), however most of the drug is bound up within the solubilizer with little available as the free drug species. Since the concentration is lower than the elevated equilibrium solubility (Figure 3a), such formulations are also not prone to precipitation. Formulation development is generally straightforward - determine the amount of solubilizer necessary to dissolve the intended dose. Membrane permeation experiments may also be helpful in determining the appropriate amount of solubilizer to employ.
 Figure 3. Concentration Profiles for Solubilized and Versus Supersaturating Systems Versus a Suspension
Figure 3. Concentration Profiles for Solubilized and Versus Supersaturating Systems Versus a SuspensionSupersaturation on the other hand is a much more complex phenomenon resulting from concentration exceeding the equilibrium solubility (Figure 3b). This comprises two stages which has been aptly described as a “spring and parachute” analogy18. The supersaturated state is generated by transferring the compound from a high energy environment (ie. through pH adjustment, a cosolvent solution or conversion to an amorphous phase) to the lower energy aqueous environment via a pH, solvent or solid form shift respectively19-21. Once created, this kinetically unstable state must also be maintained, either via delayed or prolonged precipitation. Furthermore, the precipitated material itself may also be solubility enabling if rapidly redissolving small particles or a more soluble amorphous form are generated.
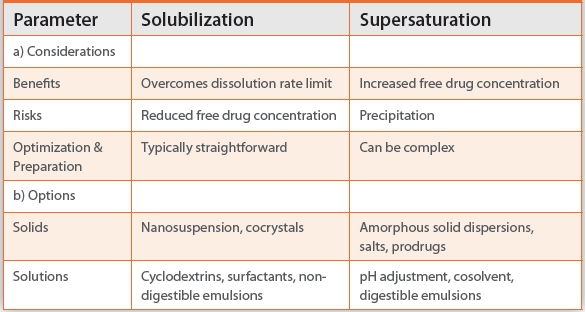
Overall, these two strategies are complimentary, each possessing unique benefits and risks (Table 1). As such they should be evaluated in parallel for promising yet solubility starved drug candidates, as illustrated in the case studies below.
Table 1. Formulation Strategy Selection Criteria
Case Study #1 - BMS-A
BMS-A is a fairly lipophilic compound with relatively low biorelevant solubility (Table 2). Two solubility enabling formulations were developed:
i) a solubilized aqueous solution containing 20% sulfobutyletherbeta- cyclodextrin
ii) a supersaturating amorphous spray-dried dispersion suspension using HPMC-AS-MG as the polymeric drug carrier at a 25% drug load
Table 2. Example Compound Characteristics

Oral dosing at 5 milligrams per kilogram (mpk) in rats demonstrated a two fold increase in exposure from the spray dried dispersion (SDD) relative to a microsuspension (Figure 4). This exposure advantage was maintained through dose escalation to 30mpk. Oral exposure from the cyclodextrin solution at 5mpk in rats was equivalent with the microsuspension and therefore initially appeared to provide no advantage. However the performance of the cyclodextrin solution was dose dependent. At 10mpk, the solution exposure was intermediate between that of the SDD and the microsuspension. At 30mpk, the solution exposure was equivalent with the SDD and continued to linearly dose escalate to 100mpk.
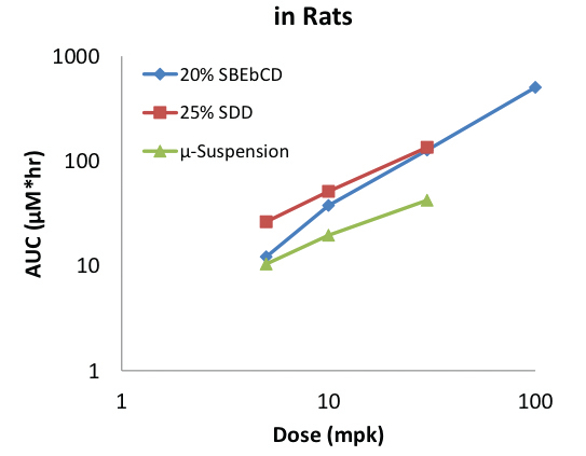
Figure 4. BMS-A Oral Exposures in Rat Versus Formulation and Dose
This example demonstrates that solubilized formulations can provide sufficient exposures so long as the amount of solubilizer is not excessive relative to the dose. In fact, solubilized formulations may be preferred over spray dried dispersions, especially earlier in the drug discovery process since they do not require specialized equipment and can be prepared under shorter timelines.
Case Study #2 - BMS-B
BMS-B is a lipophilic and crystalline compound, with relatively low biorelevant solubility (Table 2). Three enabling formulations were developed:
i) a solubilized aqueous solution with 10% hydroxypropyl-betacyclodextrin
ii) a combination supersaturating/solubilizing PEG based solution containing TPGS as a precipitation inhibitor
iii) a supersaturating amorphous spray-dried dispersion suspension using HPMC-AS-MG as the polymeric drug carrier at a 25% drug load
Oral dosing in rats at 2mpk with a 95:5 PEG400/vitamin E TPGS solution provided twice the exposure of that from a crystalline microsuspension (Figure 5). In contrast, exposure from the SDD suspension was only marginally greater than the microsuspension. Increasing the dose to 5mpk gave equivalent exposures from the cyclodextrin solution, SDD and a 70:30 PEG400/vitamin E TPGS solution. The TPGS content was increased as a precipitation prevention measure, which also resulted in a decrease in exposure relative to the solution formulation containing less TPGS despite the increased dose. At even higher doses the exposures increased for both the SDD suspension and the 70:30 PEG400/vitamin E TPGS solution, albeit to a greater extent from the solution.
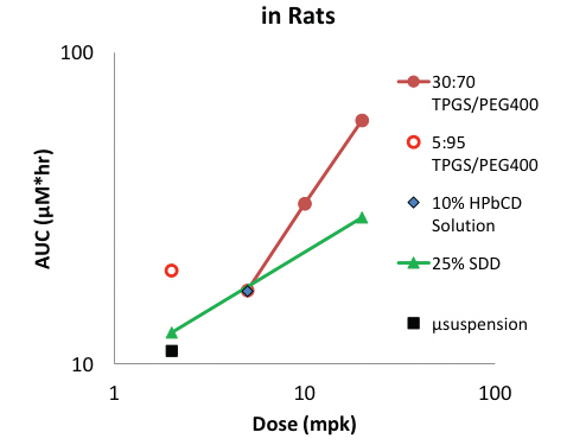 Figure 5. BMS-B Oral Exposures in Rat Versus Formulation and Dose
Figure 5. BMS-B Oral Exposures in Rat Versus Formulation and DoseThis example highlights that precipitation inhibiting excipients such as TPGS can also have solubilizing effects. The proportion of solubilization versus supersaturation depends upon the relative excipient and drug concentrations. This example also indicates the benefit of different types of supersaturating formulations. Although amorphous solid dispersions such as SDDs often provide sufficient exposure, they are not necessarily superior to other options.
Case Study #3 - BMS-C
BMS-C is a lipophilic and crystalline compound with low biorelevant solubility (Table 2). Three different formulations were developed:
i) a “solubilizing” nanosuspension, with reduced particle size enhancing dissolution rate
ii) a supersaturating PEG based solution with 5% Solutol HS-15 as a precipitation inhibitor
iii) a supersaturating amorphous spray-dried dispersion suspension using HPMC-AS-MG as the polymeric drug carrier for a 25% drug load
Oral exposures at 3mpk in rats demonstrated an improvement from particle size reduction, with the nanosuspension increasing exposure 9 fold relative to a crystalline microsuspension (Figure 6). Additionally the 95:5 PEG400/Solutol HS-15 solution and SDD suspension increased exposures approximately 2 to 3 fold over the nanosuspension in both rat and dog.
 Figure 6. BMS-C Oral Exposures in Rat and Dog Versus Formulation
Figure 6. BMS-C Oral Exposures in Rat and Dog Versus FormulationFurthermore both a PEG solution and SDD suspension demonstrated dose escalating exposures (Figure 7). However the performance of each was distinctly different once the delivery limit was reached. Exposures from the SDD suspensions essentially plateaued above 100mpk consistent with a maximum dissolution or concentration limit. The exposure from the PEG solution however decreased significantly above 30mpk which is consistent with precipitation caused by exceeding the supersaturation limit. This later example demonstrates that under some circumstances an increase in dose can actually lead to lower exposures.
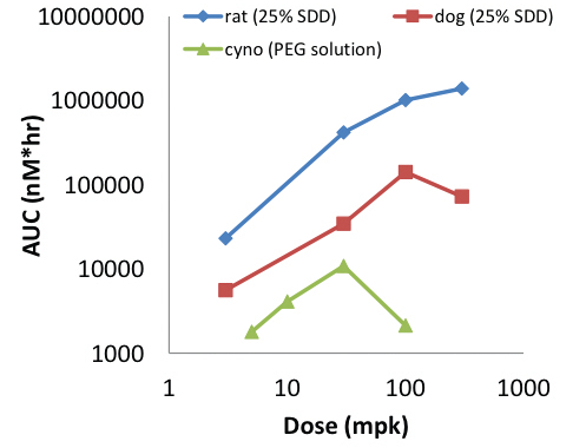 Figure 7. BMS-C Dose Escalating Oral Exposures in Rat, Dog and Cyno Versus Formulation
Figure 7. BMS-C Dose Escalating Oral Exposures in Rat, Dog and Cyno Versus FormulationSummary
Many contemporary small molecule drug candidates are poorly soluble thereby limiting their oral absorption. This limitation can be overcome through the use of solubility enabling formulations. Such formulations need to perform as intended in the dynamic and complex gastrointestinal environment which recommends phase appropriate modeling rather than relying on simple static solubility measurements.
Solubility enablement strategies fall into two categories: solubilization and supersaturation. Solubilization overcomes dissolution rate limitations but can reduce oral exposures when excessive amounts of solubilizer are employed. Supersaturation overcomes both dissolution and low inherent solubility limitations. However, supersaturating systems are by nature kinetically unstable and the absorption enhancement can be negated by precipitation. Overall, these two strategies are complimentary and valuable additions to the pharmaceutical scientist’s drug delivery toolkit.
References:
- Patrick Augustijns, Benjamin Wuyts, Bart Hens, Pieter Annaert, James Butler, Joachim Brouwers, A review of drug solubility in human intestinal fluids: Implications for the prediction of oral absorption, European Journal of Pharmaceutical Sciences 57 (2014) 322–332.
- L Di , P Fish, T Mano, Bridging solubility between drug discovery and development, Drug Discovery Today Volume 17, Issues 9–10, (May 2012), Pages 486–495
- Ashim Mitra, Chi H. Lee, Kun Cheng, Advanced Drug Delivery, John Wiley & Sons, Aug 26, 2013.
- BCS solubility - website: http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ucm128219.htm
- Kararli T, Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals, Biopharm. Drug Dispos., 16, 5, 1995.
- G Hatton, V Yadav, A Basit and H Merchant, Animal Farm: Considerations in Animal Gastrointestinal Physiology and Relevance to Drug Delivery in Humans, Article first published online: 24 FEB 2015
- Boni J, Brickl R, Dressman J, and Pfefferle M. Instant FaSSIF and FeSSIF - Biorelevance Meets Practicality. 2009. Diss Tech. 16 (3) 41-45
- Morrison J, Nophsker M, Haskell R. 2014. A Combination Turbidity and Supernatant Microplate Assay to Rank Order the Supersaturation Limits of Early Drug Candidates. J Pharm Sci. 2014 Oct;103(10):3022-32.
- Yamashita T, Ozaki S, Kushida I. 2011. Solvent shift method for anti-precipitant screening of poorly soluble drugs using biorelevant medium and dimethyl sulfoxide. Int J Pharm. 419(1-2):170-4.
- Gu C, Rao D, Gandhi R, Hilden J, Raghavan K. 2005. Using a Novel Multicompartment Dissolution System to Predict the Effect of Gastric pH on the Oral Absorption of Weak Bases with Poor Intrinsic Solubility. J Pharm Sci. 94(1): 199-208.
- Mathias N., Xu Y, Patel D, Grass M, Caldwell B, Jager C, Mullin J, Hansen L, Crison J, Saari A, Gesenberg C, Morrison J, Vig B, Raghavan K., Assessing the risk of pH-dependent absorption for new molecular entities: a novel in vitro dissolution test, physicochemical analysis, and risk assessment strategy, Mol Pharm, 10/11, 4063, 2013
- Borbás E, Balogh A, Bocz K, Müller J, Kiserdei É, Vigh T, Sinkó B, Marosi A, Halász A, Dohányos Z, Szente L, Balogh GT, Nagy ZK., In vitro dissolution-permeation evaluation of an electrospun cyclodextrin-based formulation of aripiprazole using μFlux, Int J Pharm, 491, 180, 1, 2015
- Richard Barker, Bertil Abrahamsson and Martin Kruusmägi, Application and Validation of an Advanced Gastrointestinal In Vitro Model for the Evaluation of Drug Product Performance in Pharmaceutical Development, J Pharm Sci, 103, 3704, 2014
- Alexander Alex, C. John Harris, Dennis A. Smith, Attrition in the Pharmaceutical Industry: Reasons, Implications, and Pathways Forward, John Wiley & Sons, Dec 2, 2015
- Jonathan M. Miller, Arik Dahan, Predicting the solubility–permeability interplay when using cyclodextrins in solubility-enabling formulations: Model validation, International Journal of Pharmaceutics 430 (2012) 388– 391
- Miller JM, Beig A, Krieg BJ, Carr RA, Borchardt TB, Amidon GE, Amidon GL, Dahan A., The solubility-permeability interplay: mechanistic modeling and predictive application of the impact of micellar solubilization on intestinal permeation, Mol Pharm. 2011 Oct 3;8(5):1848-56.
- Arik Dahan, Avital Beig, Viktoriya Ioffe-Dahan, Riad Agbaria, and Jonathan M Miller, The Twofold Advantage of the Amorphous Form as an Oral Drug Delivery Practice for Lipophilic Compounds: Increased Apparent Solubility and Drug Flux Through the Intestinal Membrane, AAPS J. 2013 Apr; 15(2): 347–353.
- Brouwers J, Brewster M, Augustijns P. 2009. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? J Pharm Sci. 98(8):2549-72.
- Gu C, Rao D, Gandhi R, Hilden J, Raghavan K. 2005. Using a Novel Multicompartment Dissolution System to Predict the Effect of Gastric pH on the Oral Absorption of Weak Bases with Poor Intrinsic Solubility. J Pharm Sci. 94(1): 199-208.
- Yamashita T, Ozaki S, Kushida I. 2011. Solvent shift method for anti-precipitant screening of poorly soluble drugs using biorelevant medium and dimethyl sulfoxide. Int J Pharm. 419(1-2):170-4.
- Ozaki S, Minamisono T, Yamashita T, Kato T, Kushida I.. 2012. Supersaturation-nucleation behavior of poorly soluble drugs and its impact on the oral absorption of drugs in thermodynamically high-energy forms. J Pharm Sci. 101(1):214-22.