Abstract
As the third part and final part of this series, this article focuses on future directions and challenges faced by the USP excipient Up to-Date initiative with regard to excipients. The article will provide the key findings of the USP Excipients Workshop held in November 2015 that focused on several topics, including challenges involved with introducing new or missing NF excipient monographs and opportunities to update USP excipient standards used in injectable and biologic dosage forms. Excipients are often complex substances that, within their definition and specification in a USP monograph, can exhibit relatively wide variation in composition. This article also explores the challenges involved in updating current monographs to improve characterization of excipient composition and thereby assure that impurities, concomitant components, and certain allowed additives are properly accounted for and controlled. Additionally, excipient physical/chemical properties not identified in an excipient monograph might be considered critical material attributes (CMA) to assure consistent and reliable excipient performance within the context of use in a drug product.1 Proper control of CMA may improve selection of the right excipient for the intended use and reduce the risk to drug product quality caused by excipient variability. This article also discusses excipient nomenclature inconsistencies between USP and the United States (U.S.) Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER) Inactive Ingredients Database (IID), USP excipient standards for co-processed excipients, and excipients used as atypical actives. This article also provides an overview of official USP General Notices and standards that support current excipient monograph standards and possible opportunities for developing chapters that could help drug makers gain a better understanding of excipient composition, impurities, and physical/chemical properties that may impact variability to help in selection of the right excipient. The article concludes with information on how to submit a Request for Revision to USP.
Introduction
The United States Pharmacopoeia – National Formulary (USP–NF) currently provides tests, procedures, and acceptance criteria that help to ensure the quality and safety of excipients used in drug products and dietary supplements marketed in the U.S. The standards in USP–NF are also recognized globally, as they are used worldwide. The USP–NF defines pharmaceutical excipients as substances other than the active pharmaceutical ingredient (API) that have been appropriately evaluated for safety and are intentionally included in a drug delivery system.2 Excipients are components of the drug product intentionally added to enable the delivery, ease of manufacture, and stabilization of the API in a formulation. Excipients are listed in the IID,3 which provides information on previously accepted excipient levels and routes of administration in drug products approved by FDA. There is no independent regulatory approval process for excipients outside of an FDA drug product review. This leads to challenges associated with the introduction of new excipients in drug development and into USP-NF.
Challenges and Opportunities in Development of New Excipient Monographs in NF
The attendees at the USP Excipient Workshop discussed the growing need for new excipients to support innovative drug development, particularly in the realm of new delivery systems and stable novel dosage forms. Unlike new chemical entities which provide breakthrough therapies in the API world, discovery and manufacturing of a new chemical entity is almost never a feature of a new excipient. There are many ways to create a new excipient. FDA may consider any substance not listed in IID to be a new excipient, and as discussed below, inconsistent nomenclature can make an excipient already listed in IID appear to be new. A new excipient can alternatively be obtained from excipients having a prior and repeated history of use by, for example, co-processing a combination of established excipients, and thus creating a new modification of the physical form or a new variation of the excipient composition. Such modifications often raise the question as to whether either a new USP excipient monograph should be developed or an existing monograph should be modified.4 There is an opportunity for new excipients to meet the ever-increasing challenges drug development scientists and engineers face in the delivery of an active pharmaceutical ingredient (API) and manufacture of a drug product. However, amid concerns about FDA acceptance of a new excipient, pharmaceutical manufacturers stay within the boundaries for excipient levels and routes of administration listed in the IID.5,6 This conundrum cannot be addressed without considering a new excipient’s safety profile. For excipients that are currently listed in the IID, the USP website www.usp.org contains a list of priority missing monographs for development, with input requested from sponsor(s) and a donor recognition program.7-9 Classes of new excipients are described by Moreton10 to include:
- new chemical material;
- co-processing of existing material;
- new semi-synthetic derivatives or new chemistry (e.g. degree of substitution) of existing materials;
- excipients used in food (e.g. Generally Recognized As Safe (GRAS) database listed materials) and animals, now proposed for pharmaceuticals;
- excipients for new route of administration;
- new botanical source or manufacturing process for existing materials;
- new physical grades.
Challenges identified by USP include prioritizing monographs that need updating, defining meaningful specifications and obtaining procedures and acceptance criteria from sponsors to support a new route of administration. USP welcomes opportunities to work together with FDA and excipient users and manufacturers to expedite the development of monographs. USP will also work independently with the European Pharmacopoeia (EP) and Japanese Pharmacopoeia (JP) in development of harmonized monographs for new excipients.
Understanding USP’s Role with Excipients Used in Biologics
In particular, the topic of new excipients for biologic dosage forms has drawn more attention recently because of the unique challenges involved in formulating and stabilizing drug substances exhibiting higher-order chemical structure that is necessary for activity. Owing to poor stability, release and absorption via the gastrointestinal tract for biologic drug substances and biologics, they are most commonly delivered parenterally as sterile products. The latter are often produced using non-sterile excipients. Thus, it is desirable to control the microbiological quality of excipients at a level compatible with the biologic manufacturing process. However, the retention of structural integrity of a biologic drug substance could conceivably be affected by the composition of excipients or by excipient impurities. Questions for discussion at the workshop included:1) Should USP have lower limits on bioburden in specifications for all parenteral use, aiming to minimize the potential for excipients to contribute to higher endotoxin levels?; and 2) Should USP develop monographs or a general chapter(s) to help in the selection of the excipient best suited for parenteral use? Stakeholder collaborations can stimulate additional avenues to both update and harmonize excipients used in injectable biologics.
FDA’s Inactive Ingredient Database
The USP Excipient Workshop discussed the FDA’s Federal Register Notice regarding the enhancement of the utility and usability of the IID to help the agency identify and ultimately establish best practices and issue a technical guide or draft guidance. At the workshop, attendees discussed the current challenges with the IID and the importance of avoiding contradictory nomenclature for excipients.11
USP has a long-standing legal role in establishing the names of drugs, including excipients, under Section 502(e)(3) of the Federal Food, Drug and Cosmetic Act. Under this provision, the established name (nonproprietary name) of a drug or component is the official title used for the drug or component in an official compendium such as USP or NF (USP–NF), unless FDA has designated another name via regulation. Thus, workshop attendees seemed to agree that when a USP–NF standard exists, the excipient would ideally appear in IID under the offiial title established in the USP–NF. However, this is not always the case.
A major effort is underway at FDA to rectify current entries in the database and to provide the means going forward to improve the quality and utility of data. However, many excipients with new or revised monographs in the current USP work plan still use nomenclature that traces to non-pharmaceutical use. For example, excipients used in topical drugs are generally marketed using cosmetic naming convention. The FDA Substance Registration System (SRS) provides the preferred name as well as synonyms for excipients which can help answer questions such as whether an excipient is considered new. Thus, USP compendial nomenclature could be a source of preferred names, and applicants can assist by using preferred names in the composition statement section of the application. For excipients that are non-compendial, SRS can be accessed to find the preferred name for the composition statement.
Co-Processed Excipients
A co-processed excipient contains more than one excipient whereby co-processing does not involve formation of a chemical bond between any of the individual excipients. The USP Excipients Expert Committee (Excipients EC) deemed it appropriate to include coprocessed excipients in the NF because these excipients possess certain physical characteristics that are different from or altered from their corresponding simple physical admixtures. Co-processed excipients should follow proposed criteria established to define when USP would consider advancing a prospective new monograph for a co-processed excipient into NF. These criteria were developed to differentiate multi-component co-processed excipients from the more commonly encountered excipients that have been the norm in NF. Due to the complex processes used to manufacture multi-component excipients, their proprietary nature, and the lack of comprehensive understanding of these types of excipients, the EC proposes to limit the current criteria to solid co-processed materials. A request for revision (RFR) to develop a co-processed excipient monograph should consider the criteria appearing in the Stimuli article on Co-processed Excipients.12
When the excipient is submitted as a potential NF monograph, information relating to its quality must meet current NF submission requirements.13
Atypical Active Ingredients
USP also contains several monographs for excipients used in drugs as active ingredients, that are termed atypical actives or dual-use drug ingredients. Monographs for excipients used as atypical actives are typically published in the USP section of the USP–NF with the title listed in the NF section, for example, “Glycerin – see Glycerin General Monograph), indicating that it is used as both an excipient and active ingredient. Labeling to indicate compliance with either USP or NF (or both) could signal the intentions of manufacturers regarding how an ingredient was designed to be used. This is an important distinction because an excipient’s use as an API generally leads to the most stringent ingredient Good Manufacturing Practice (GMP) expectations.
Excipient monographs for atypical active ingredients are also part of the USP Up-to-Date initiative, with a primary focus on replacing nonspecific identification tests with those suitable for compliance with regulatory requirements for excipient identity. An equally important goal for monograph development is replacement of non-specific assay methods and addition of analytical methods that allow better measurement of the purity of excipients used as active ingredients. Polyethylene Glycol (PEG) 3350 USP is an excellent example. PEG 3350 was originally included in the PEG NF monograph as one of several grades of the excipient. PEG NF does not contain an identification test and the assay measures average molecular weight. The Excipient EC worked with stakeholders to develop a separate PEG 3350 USP monograph by proposing appropriate test procedures and methodologies in Pharmacopeial Forum (PF) 39(6) and PF41(4) to uniquely identify PEG 3350 and to properly determine the product’s strength (content).
Impact of Excipient Variability on Drug Product
Excipients are known to exhibit variability in their critical material attributes (CMAs) due to various factors associated with excipient manufacturing.14 Because generally excipients are added in fixed amounts when products are manufactured, such variability could translate into product and process variation. The identification and evaluation of excipient CMAs hold promise for the development of novel control strategies that could lead to improved quality and manufacturing capability throughout a product’s life-cycle. The USP Excipient Workshop included a presentation about the National Institute of Pharmaceutical Training and Education (NIPTE)/FDA PharmaHub database,15 a unified attempt to catalog data from testing of commercial excipient batches, showing how excipient physical/chemical properties may vary. Not all performance-related CMAs of an excipient may be identified or evaluated by specific tests and Acceptance Criteria" listed in compendial monographs.14 As stated in General Notices 4.10. Monographs, because monographs may not provide standards for all relevant characteristics, some official substances may conform to the USP or NF standard but differ with regard to nonstandardized properties that are relevant to their use in specific preparations.16 To provide general information about performance-related tests linked to specific uses of excipients, USP has developed General Chapter Excipient Performance.17
USP chapteris designed to provide an overview of typical excipient material attributes for many functional categories along with additional tests that may be useful in evaluating excipient attributes that are not typically included in compendial monographs. Selection of the appropriate tests and specifications that are necessary to ensure consistent and reliable excipient performance requires an understanding of the formulation and manufacturing processes, the dosage form performance requirements and the physical and chemical properties of each ingredient in the dosage form. The chapter provides references to standard USP–NF test procedures that can be used by both manufacturers and users. Provided USP chapteris not referenced in an official excipient monograph, any tests performed in accordance with this chapter are considered non-mandatory as far as compliance with the excipient monograph in question. The chapter does not impose limits or specifications since the properties of an excipient that are required will vary and depend upon the product, manufacturing process, quantity, and intended function. Methods fromand corresponding acceptance criteria can be referenced in a drug application. In such an instance, methods that reference USP chapterand corresponding acceptance criteria for the excipient might be considered part of the specification for the manufacturing of the particular product that is the subject of a drug application.
Ongoing Challenges to the Excipient Up-to-Date Initiative
The workshop attendees discussed the importance of determining how to best ensure that each excipient monograph reflects the quality of excipients currently used in drugs on the US market and the extent to which monographs can help promote excipient integrity (authenticity and fitness for purpose). There is also a need to understand excipient composition and evaluate how to define and address impurities as opposed to concomitant components. A brief overview was provided on how the USP Up-to-Date initiative will impact excipient monographs. In the short term, efforts of the Excipient EC will be focused on developing specific identification tests for those excipient monographs currently lacking them. The Excipient EC will consider how to best approach analytical methods that allow better characterization of composition of excipients. FDA liaisons participate in compendial excipient monograph development to the extent possible in fulfillment of the Agency’s role in promoting development of pubic standards which in turn promote pharmaceutical quality and patient safety. The workshop identified possible opportunities for developing additional general chapters that could help drug makers gain a better understanding of excipient composition (impurities, physical/chemical properties that may impact variability) to select the right excipient.
Submitting a Request for Revision to USP
The purpose of a Request for Revision (RFR) generally is to create a new monograph for a new excipient or to revise or update an existing monograph; note that every revision must be meaningful, add value to the public standard, and contribute to the public health.13 USP is actively engaged in updating official USP–NF monographs that currently utilize outdated technology, have safety/environmental concerns, or are missing procedures for key attributes such as identification, assay, and impurities, with a goal of improving characterization of the excipient.
Table 1. Request for Revision for an NF excipient monographthe RFR typically includes the following sections (not every excipient monograph will contain every section)
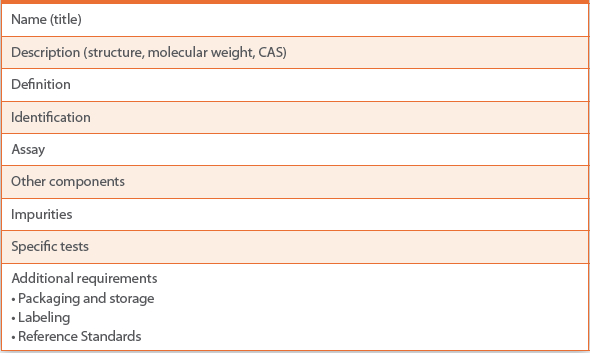
An RFR for an USP–NF excipient monograph justifies the specification and typically includes the following sections (not every excipient monograph will contain every section): name (title), description (structure, molecular weight, CAS), definition, identification, assay, other components, impurities, specific tests, and additional requirements (packaging and storage, labeling, reference standards) (see Table 1). The Checklist for Submitting Requests for Revision to the USP–NF is available on the USP website.13
Conclusions
USP–NF excipient monographs need to be Up-to-Date to provide maximum benefit to the FDA, manufacturers and users of excipients. Monographs for excipients should be updated periodically to reflect current understanding regarding the role of excipients in new drug formulations and function in the manufacturing process, and because of the ever-expanding use of excipients. The control of impurities that may contribute to deteriorating product quality, the addition of a test that can reveal the composition of an excipient and the inclusion of permitted additives preventing product degradation are key examples of how the monograph can play a critical role in defining the minimum standard of excipient quality.
Monograph development can help meet increasing and newly identified challenges faced in the formulation of drugs and drug delivery systems including stabilization of the higher-order structure of active substances in biologics. Naming of excipients using the official name can be integrated into a future version of IID to eliminate confusion for IID users. General chapters on excipient composition could allow all stakeholders to more readily distinguish between impurities and concomitant components. General chapters could also help drug developers gain a better understanding of excipient composition (impurities, physical/chemical properties that may impact variability) thereby helping select the excipient best suited for the intended purpose.
References
- FDA Guidance for Industry Q8 (R2) Pharmaceutical Development http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073507.pdf
- USP 38 – NF 33 (United States Pharmacopeial Convention) General Information Chapter, Good Manufacturing Practices for Bulk Pharmaceutical Excipients, Rockville, MD: USP; 2015.
- FDA CDER Inactive Ingredient Database (IID) http://www.accessdata.fda.gov/scripts/cder/iig/index.cfm accessed December 26, 2015
- S. Ali, Challenges and Opportunities in Development of Novel Excipients & Monographs in USP, Presentation in USP 2nd Excipients Workshop, Nov. 17-18, 2015.
- FDA CDER/CBER Guidance for Industry Nonclinical Studies for the Safety Evaluation of Pharmaceutical Excipients 2005 http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079250.pdf Accessed on January 20, 2016
- K. Otilia, Application Challenges and Examples of New Excipients in Advanced Drug Delivery Systems, American Pharmaceutical Review, Mar. 01, 2011
- USP Priority New Monograph Items http://www.usp.org/usp-nf/development-process/priority-new-monographs Accessed December 26, 2015
- USP List of missing excipients accessed December 26, 2015 http://www.usp.org/usp-nf/ development-process/priority-new-monographs/list/excipients
- USP Donor recognition program Accessed December 26, 2015 http://www.usp.org/uspnf/development-process/donor-recognition-program USP website missing monographs
- R. Moreton, Functionality and Performance of Excipients in a Quality-by-Design World, Part IX: New Excipients. American Pharmaceutical Review Apr 34-37 (2010).
- USP comments regarding the enhancement of the utility and usability of the Inactive Ingredient Database [Docket No. FDA–2015–N–2986] http://www.regulations.gov.
- Stimuli to the Revision Process: USP Responses to Comments on Stimuli Article: Coprocessed Excipients, PF 37(3) [May–June 2011].
- USP Monograph Submission Guideline - Submitting Requests for Revision to the USP–NF http://www.usp.org/usp-nf/development-process/submit-new-monographs/submissionguidelinesAccessed December 26, 2015
- G. Amidon, C. Sheehan, Compendial Standards and Excipient Performance in the QbD era: USP Excipient Performance Chapter <1059>, American Pharmaceutical Review, 2011.
- The NIPTE-FDA Excipients Knowledge Base https://pharmahub.org/ accessed on December 27, 2015.
- USP 38 – NF 33 (United States Pharmacopeial Convention) General Notices, 4.10 Monographs. USP, Rockville, MD, USA, 2015
- USP 38 – NF 33 (United States Pharmacopeial Convention) General Information Chapter, Excipient Performance s <1059>, Rockville, MD: USP; 2015.