The importance and value of modular continuous processing technologies to provide economic and sustainability advantages means that the industry is able to adapt more rapidly to changing market demands. Factors other than scientific are the barriers to change from batch to continuous production. The bio/pharmaceutical industry has reached a stage that requires a change in the production paradigm. For certain classes of biopharmaceutical products upstream continuous manufacturing has always been applied: for example, unstable proteins that rapidly degrade in the culture broth. In order to obtain a high quality product, the residence time in the reactor must be minimized. This can only be achieved with continuous cultivation and preferably with perfusion reactors.
Furthermore, this is a universal production platform that can be extended to other classes of products, such as antibodies, which are relatively stable molecules. Continuous manufacturing is as productive and with a much smaller footprint of the manufacturing plant, avoiding multiple non-value added unit operations. In essence, the investment for a continuous plant is much smaller compared to a batch-operated one.
Current Regulatory Point of View
The current regulatory environment supports advancing Regulatory Science and Innovation, which may include abandoning some traditional manufacturing practices in favor of a cleaner, flexible, closed system, for more efficient continuous manufacturing. Regulatory authorities in the three ICH regions and beyond are encouraging industry to adopt new technology as supported by ICH Q8(R2), Q9, Q10 and Q11, along with the introduction of Quality by Design (QbD) concepts, emphasizing science and risk-based approaches to assure product quality. The regulatory expectations for assurance of reliable and predictive processing that is technically sound, risk-based, and relevant to product quality in a commercial setting are the same for batch and continuous processing.
ICH Guidelines: The emergence of ICH Q8 (R2), Q9, Q10, and Q11 guidelines and accompanying ICH Q-IWG Points to Consider and Q&A documents emphasized that a prospective science and risk-based approach to development and lifecycle management could increase the assurance of quality of pharmaceutical products. Collectively, these guidelines reinforced the adoption of risk–based [ICH Q9], systematic and science–based approaches [ICH Q8(R2) and ICH Q11], and a robust pharmaceutical quality system [ICH Q10], to establish an increased level of process understanding and product knowledge.25
US FDA Guidelines: The FDA Guidance for Industry PAT,26 A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance specifically identifies that the introduction of continuous processing may be one of the outcomes from the adoption of a scientific risk-based approach to process design. Process understanding, control strategies, plus on-line, in-line, or at-line measurement of critical quality attributes provide for control strategies that include real time quality evaluation that is at least equivalent to, or better than, laboratory-based testing on collected samples.
On Process Validation/Continual Verification: FDA Guidance on Process Validation/Continual Verification aligns process validation activities with a product lifecycle concept. The guidance encourages the use of modern pharmaceutical development concepts, quality risk management, and quality systems at all stages of the manufacturing process lifecycle. The lifecycle concept links product and process development, qualification of the commercial manufacturing process,27 and maintenance of the process in a state of control during routine commercial production. This guidance supports process improvement and innovation, including continuous manufacturing, through sound science.
EU Guidelines: The ICH Guidelines referenced above apply in the European Union. EU Guidelines that might be particularly relevant to continuous manufacturing include the Guidelines for Process Validation where the concept of continuous process verification is introduced; the Guideline on NIR29 as it is often used as a Process Analytical Technology (PAT) tool for process monitoring and/or control, and the Guideline on Real Time Release Testing.30 Although not required, continuous manufacturing is commonly coupled with Real Time Release Testing (RTRT).
L. Yu, PhD., Deputy Director FDA, states; “Today, a new and exciting technology — continuous manufacturing — enables much faster production and more reliable products through an uninterrupted process. How much faster is continuous manufacturing? In some cases, manufacturing that takes a month by batch technology might only take a day using continuous manufacturing techniques.”1
And the article ends with; “FDA will continue our efforts to encourage the advancement of continuous manufacturing as one of a variety of ways to enhance the quality of the medications used by the American public.”1
FDA Allows First Switch From Batch to Continuous Manufacturing for HIV Drug; Janssen’s HIV-1 treatment Prezista (darunavir), the FDA Approved the manufacturing change over, April 9, 2016.2
Vertex’s cystic fibrosis drug Orkambi [lumacaftor/ivacaftor] has been using the continuous manufacturing process since its approval in July 2015.2
In 2015, Aprecia’s first 3D printed drug product was approved by the FDA (Spritam® (levetircetam) tablets for oral suspension). Apricia manufacturing model is through exclusive licensing of the pharmaceutical applications of the MIT three-dimensional printing (3DP) process.
In biologics continuous manufacturing: Actual use of perfusion for commercial manufacture is more widespread, but bioprocessing manufacturers are becoming increasingly reticent to publicly share details about their processes. Manufacturers such as Genzyme, Bayer, Janssen, Merck-Serono, Novartis, and Lonza for Eli Lilly manufacture approximately 19 marketed recombinant protein and monoclonal antibody (mAb) products using perfusion or elements of continuous processing, and these products are predominantly blockbusters with annual revenues totaling ~US$20 billion.3
Conventional Batch Manufacturing (Figure 1, and 2)
 Figure 1. Batch process manufacturing from seed to unit dose
Figure 1. Batch process manufacturing from seed to unit dose Figure 2. Batch Tablet Manufacturing. Product collected after each unit operation, Finished product is tested off-line, after processing is complete, and Actual processing time; days to weeks.
Figure 2. Batch Tablet Manufacturing. Product collected after each unit operation, Finished product is tested off-line, after processing is complete, and Actual processing time; days to weeks.The traditional batch manufacturing is gone through every possible improvement step to optimize the process, decrease complexity, and reduce Capital Expenditure (CapEx) and Operational Expense (OpEx) that could improve and streamline the biomanufacturing of drug substance and product delivery to patients. The industry implemented many of the available technologies and operational management tools to achieve this task;
- Increase “Overall Asset Effectiveness” (OAE)
- Reduce “Throughput Time” (TPT)
- Improve “Quality” (Q) by using seamlessly integrated and well characterized processes
Implementing Lean Manufacturing and other optimization programs to improve traditional batch biomanufacturing resulted in some improvements but the industry is still struggling with;
- Operation
- Major “efficiency gains” have been implemented and
- Additional “quantum leap efficiency gains” are unlikely
- “Manufacture and ship” across continent
- Compliance
- Many “manual” checks
- Deviations and investigations
- Difficult “root cause” analysis
- Quality
- Reliance on “in-process” and “end-product” testing The Mainstreaming of Continuous Flow API Synthesis
The pharmaceutical industry is moving toward commercial-scale continuous processes for small-molecule (API) manufacturing. Continuous processing for the production of intermediates for Smallmolecule (APIs) is no longer viewed as a technology of the future. Most pharmaceutical companies with in-house manufacturing use flow chemistry to some extent, and smaller companies that outsource production expect their contract manufacturing partners to have continuous-flow systems. Advances in microreactor technology for commercial-scale production and implementation of continuous downstream processes will ultimately enable the complete continuous synthesis of complex organic molecules required for small-molecule API manufacturing. Widespread adoption, however, will occur slowly as the industry shifts from the infrastructure in place in today’s batch manufacturing, as shown in Figures 1 and 2, to smaller, modular, and flexible facilities, as shown in Figures 3 and 4.
 Figure 3. Chemical (small molecule) continuous manufacturing process “Blue Sky”, from raw material to tablet in one small footprint manufacturing operation.
Figure 3. Chemical (small molecule) continuous manufacturing process “Blue Sky”, from raw material to tablet in one small footprint manufacturing operation. Figure 4. Batch vs continuous manufacturing “Blue Sky”. “Blue Sky” approach, reduce the number of unit operations, reduce equipment size, and manufacture 24/7.
Figure 4. Batch vs continuous manufacturing “Blue Sky”. “Blue Sky” approach, reduce the number of unit operations, reduce equipment size, and manufacture 24/7.An economic comparison of “Blue Sky” continuous manufacturing as compared to batch manufacturing is depicted in Table 1, and reveals the benefit of the many improvements in CapEx and OpEx. Comparing Figure 3 to Figure 1, we could deduce simplification of the operations and the modularization of the system from seed to unite dose manufacture.
Table 1. Using “Blue Sky” continuous manufacturing vs batch economic comparison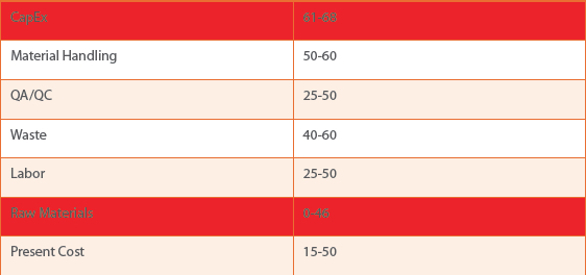
Biopharmaceutical Systems: Upstream Fed-Batch system: an overall improvement resulted due to achieving higher manufacturing titer (viable cell density) as shown in Figure 9, improved controls PAT (Process Analytical Technology), and QbD (Quality by Design), flexible equipment and system designs (utilizing single use technology where applicable) that progressed the biomanufacturing from an overall complex line shown in Figure 5, to an improved simpler operation shown in Figure 6.
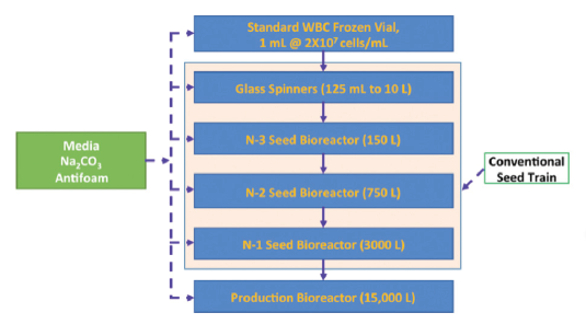 Figure 5. Typical conventional production scale fed-batch system, upstream (the production scale reactor could be up to 30 KL)
Figure 5. Typical conventional production scale fed-batch system, upstream (the production scale reactor could be up to 30 KL) Figure 6. Typical Production scale improved fed batch system, upstream (the production scale reactor could be up to 2 KL)
Figure 6. Typical Production scale improved fed batch system, upstream (the production scale reactor could be up to 2 KL)For upstream perfusion system an overall improvement resulted due to better titer, higher volumetric flowrate (volumetric productivity), as shown in Figure 10, flexible equipment and better system designs from the overall complex operation shown in Figure 7, to the simpler operation shown in Figure 8.
 Figure 7. Typical conventional production scale perfusion system, upstream
Figure 7. Typical conventional production scale perfusion system, upstream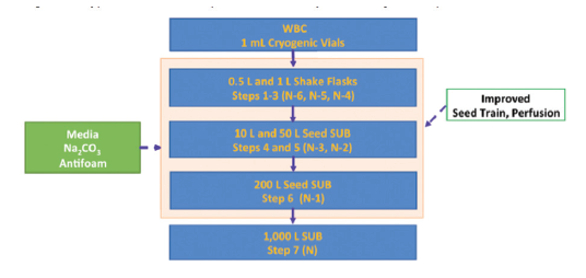 Figure 8. Typical improved Production scale perfusion system, upstream
Figure 8. Typical improved Production scale perfusion system, upstream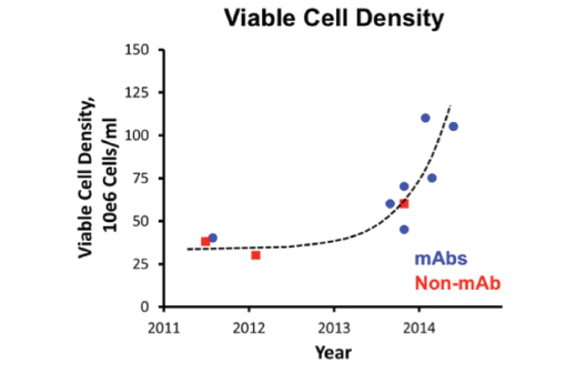 Figure 9. Viable Cell Density, Rapid progress in upstream production capability, significant upward potential24
Figure 9. Viable Cell Density, Rapid progress in upstream production capability, significant upward potential24 Figure 10. Volumetric Productivity, rapid progress in upstream production capability, significant upward potential24
Figure 10. Volumetric Productivity, rapid progress in upstream production capability, significant upward potential24Continuous Biomanufacturing
To distinguish between batch manufacturing and continuous manufacturing: in batch manufacturing, all materials are charged before the start of processing and discharge at the end of processing. In continuous manufacturing, materials are simultaneously charged and discharged from the process.
To achieve upstream continuous biomanufacturing, it requires a paradigm shift thinking and an implementation of systems that are;
- Modular (Modularity)
- Flexible (Flexibility)
- Portable (Portability)
- Compact (Scale-out, Expansibility)
- Less real estate (Small Foot-Print)
- Cost effective ($/Unit deliverable to patient)
- Optimized (Optimization)
- Friendly on change-over (Multi-Product)
- Compliant (Regulatory)
- Efficient (Efficiency)
- Sustainable (Sustainability)
This requires a design and implementation of new technologies to attain the goal as illustrated in the following example, in Table 2.=
Table 2. Upstream Continuous Manufacturing: Action required, Technology needed and Benefit gained.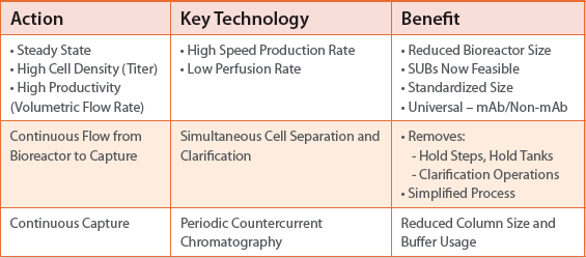
By implementing what is outlined in Table 2 and clinical experimental data (proof of concept reported by few firms that conducted the studies) the upstream system would result in the simple system as illustrated in Figure 11.
 Figure 11. Continuous biomanufacturing – upstream
Figure 11. Continuous biomanufacturing – upstreamMeasurement and control for enabling continuous processing adoption in the biotech and biopharmaceutical industries – the shift from batch to continuous production methods is transforming the future of biopharmaceutical manufacturing. The potential economic gains from increasing capacity utilization and reducing the length of process development, product release times and capital costs are driving the paradigm shift. Spurred by potential improvements to quality, patient safety and the time required for breakthrough medicines to reach patients.
The U.S. Food and Drug Administration (FDA) has advocated a move to continuous manufacturing (CM). However, with new methods come new challenges — particularly for process design, measurement, material traceability and control. Manufacturers, suppliers and research institutions are collaborating to solve these challenges on projects across the globe.
The status of continuous bioprocessing in the biopharmaceutical industry is based in part on research on how manufacturers can overcome the challenges related to quality, compliance, material traceability and process design and control.
Continuous Biomanufacturing - Upstream
A process platform for a fully integrated continuous fermentation, clarifications harvest and capture system of the desired protein consists of a stirred tank reactor with a cell retention device, Figure 11, the reactor is continuously harvested and the cell-free effluent captured by a continuous chromatographic process. In such a process, the culture supernatant is loaded to full saturation on a chromatography column and after breakthrough; the column effluent is loaded on a second column. After full saturation the column is disconnected from the second column and product is recovered, whereas the second one is loaded to full saturation. After breakthrough of the second column the effluent is loaded onto a third column. The second column is disconnected again and product is recovered. This process is also known as countercurrent chromatography and has been implemented at pilot scale. The system is more complex than a typical batch chromatography process, but fully automated and with higher productivity. The product quality could be maintained at high levels during long-term steady state operation of the integrated continuous system. In light of the current transition of the biopharmaceutical industry towards Quality-by-Design and real-time product release, continuous manufacturing technology is a big step in getting closer to this ambitious goal.
Continuous Biomanufacturing - Downstream
Downstream processing focuses on yield and productivity as well as on purity and process capacities. An increase in separation efficiency of single unit operations is achieved by expansion of existing facilities and by optimization of existing and alternative processes.5 These include the establishment of platform technologies, high-through-put methods with approaches based on QbD and DoE-based (Design of Experiment) experimental optimizations,5,13,15 additionally, an integration of modeling and simulation of unit operations as well as the use of mini-plant facilities.
Traditionally, monoclonal antibodies were purified by a sequence of different chromatographic and membrane-based operations. 5,7,8,9,10,16,17 A virus-inactivating operation, a filtration-based virus-reducing step and a final diafiltration have to be included.4,7,10,11,16,17
Continuous biomanufacturing is currently being seriously considered in the biopharmaceutical industry as the possible new paradigm for producing therapeutic proteins, due to production cost and product quality related benefits.
An example7 monoclonal antibody producing CHO (Chinese Hamster Ovary) cell line was cultured in perfusion mode and connected to a continuous affinity capture step. The reliable and stable integration of the two systems was enabled by suitable control loops, regulating the continuous volumetric flow and adapting the operating conditions of the capture process. For the latter, an at-line HPLC measurement of the harvest concentration subsequent to the bioreactor was combined with a mechanistic model of the capture chromatographic unit. Thereby, optimal buffer consumption and productivity throughout the process was realized while always maintaining a yield above the target value of 99%. Stable operation was achieved at three consecutive viable cell density set points (20, 60, and 40×106 cells/ mL), together with consistent product quality in terms of aggregates, fragments, charge isoforms, and N-linked glycosylation. In addition, different values for these product quality attributes such as N-linked glycosylation, charge variants, and aggregate content were measured at the different steady states. The amount of released DNA and HCP was significantly reduced by the capture step for all considered upstream operating conditions.
In the current environment of diverse product pipelines, rapidly fluctuating market demands and growing competition from biosimilars, biotechnology companies are increasingly driven to develop innovative solutions for highly flexible and cost-effective manufacturing. To address these challenging demands, integrated continuous processing, comprised of high-density perfusion cell culture and a directly coupled continuous capture step, can be used as a universal biomanufacturing platform.
Another example,8 reports the first successful demonstration of the integration of a perfusion bioreactor and a four-column periodic countercurrent chromatography (PCC) system for the continuous capture of candidate protein therapeutics. Two examples are presented: (1) a monoclonal antibody (model of a stable protein) and (2) a recombinant human enzyme (model of a highly complex, less stable protein). In both cases, high-density perfusion CHO cell cultures were operated at a quasi-steady state of 50–60×106 cells/ mL for more than 60 days, achieving volumetric productivities much higher than current perfusion or fed-batch processes. The directly integrated and automated PCC system ran uninterrupted for 30 days without indications of time-based performance decline. The product quality observed for the continuous capture process was comparable to that for a batch-column operation. Furthermore, the integration of perfusion cell culture and PCC led to a dramatic decrease in the equipment footprint and elimination of several non-value-added unit operations, such as clarification and intermediate hold steps. These findings demonstrate the potential of integrated continuous bioprocessing as a universal platform for the manufacture of various kinds of therapeutic proteins.
Conclusion
Traditional batch biomanufacturing is restrictive:
Inherent obstacles in batch biomanufacturing:
- Capital intensive
- Consumes large real estate
- Requires enormous time to build the facility
- Requires large staff to operate
- The demand for capacity is going up
- And there is a need for local manufacturing
Disadvantages of batch processing
- Defined batch size (output quantity driven by batch size)
- Multiple, sequential process steps, end to end
- Many interruptions between/during process steps
- Long waiting times between single process steps
- Numerous transport steps between process steps
- Lengthy throughput times from start to finish
- High levels of raw material and intermediate inventories required
- Extensive validation and scale-up activities needed
- Physical and organizational separation in operations and development
- Quality measured by in process sampling/control and end product testing
Continuous Manufacturing Platform in Summary
 Figure 12. Cell density, current process vs new platform24
Figure 12. Cell density, current process vs new platform24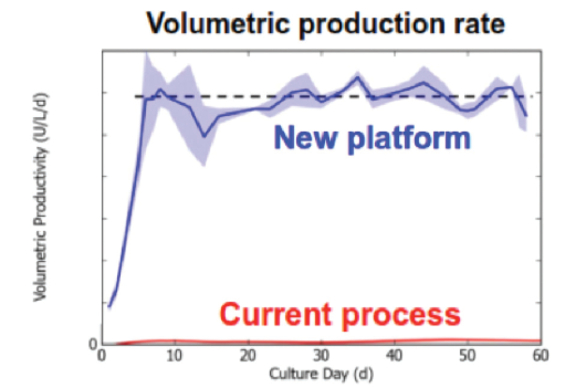 Figure 13. Volumetric production rate, current process vs new platform24
Figure 13. Volumetric production rate, current process vs new platform24 Figure 14. Going from traditional batch biomanufacturing to integrated continuous
Figure 14. Going from traditional batch biomanufacturing to integrated continuous bioprocessing
24Integrated continuous biomanufacturing, Figures 3, and 4 (Small molecule API synthesis),12, 13, and 14 (biologics manufacturing); is:
- Universal, standard platform (various proteins)
- Steady state (metabolism)
- Closed system (minimized microbial issues)
- No scale-up, same scale for pilot & manufacturing
- Due to scale, compatible with single-use/disposable technology
- Minimized hold time
- Continuous flow
- High volumetric productivity
- Integrated, modular, simplified operation
- Flexible capacity, increase and or decrease
Advantages of Continuous Manufacturing
- Integrated bioprocessing with fewer steps
- no manual handling
- increased safety
- shorter processing times
- increased efficiency
- Smaller equipment and facility footprint
- more flexible operation
- reduced inventory
- lower costs
- less work-in-progress material
- smaller ecological footprint (sustainability)
- On-line monitoring and control for increased product quality assurance in real-time
- amenable to Real-Time Release Testing approaches
- consistent quality
CAPEX and OPEX advantages of Continuous Manufacturing:
- Integration of compliance/quality within the process
- Reduction of asset footprint (40-90%)
- Reduction in CapEx (25-60%)
- Reduction of OpEx (25-60%)
- Reduction of raw material and intermediate inventories
- Flexibility in supply size
- Reduction in overall development times for drug substances and drug products and Improving time to market
- Assuring availability of high quality, safe, and efficacious drug products to patients
What we are doing brings technology and innovation to the forefront.
Continuous manufacturing (CM) and integrated continuous manufacturing (ICM) have been successfully implemented at certain drug product manufacturers with the assistance of educational institutions, equipment manufacturers, and suppliers.
References
- Lawrence Yu, Ph.D.; Deputy Director, FDA; Office of Pharmaceutical Quality, Center for Drug Evaluation and Research, Article Posted: April 12, 2016, FDA Voice.
- http://www.raps.org/Regulatory-Focus/News/2016/04/12/24739/FDA-Allows-First-SwitchFrom-Batch-to-Continuous-Manufacturing-for-HIV-Drug/#sthash.AGIwm1JQ.dpuf
- 11th Annual Report and Survey of Biopharmaceutical Manufacturing, E. Langer, . BioPlan Associates, April 2014, pp. 103-116.
- Jain, E.; Kumar, A. Upstream processes in antibody production: Evaluation of critical parameters. Biotechnol. Adv.2008, 26, 46–72. [Google Scholar] [CrossRef]
- Shukla, A.A.; Thömmes, J. Recent advances in large-scale production of monoclonal antibodies and related proteins Trends Biotechnol. 2010, 28, 253–261. [Google Scholar] [CrossRef]
- Gagnon, P. Technology trends in antibody purification. J. Chromatogr. A 2012, 1221, 57–70. [Google Scholar] [CrossRef]
- Chon, J.H.; Zarbis-Papastoitsis, G. Advances in the production and downstream processing of antibodies. New Biotechnol. 2011, 28, 458–463. [Google Scholar] [CrossRef]
- Kelley, B. Industrialization of mAb production technology: The bioprocessing industry at a crossroads. MAbs 2009, 1, 443–452. [Google Scholar] [CrossRef]
- Low, D.; O’Leary, R.; Pujar, N.S. Future of antibody purification. J. Chromatogr. B 2007, 848, 48–63. [Google Scholar] [CrossRef]
- Liu, H.F.; Ma, J.; Winter, C.; Bayer, R. Recovery and purification process development for monoclonal antibody production. MAbs 2010, 2, 480–499. [Google Scholar] [CrossRef]
- Birch, J.R.; Racher, A.J. Antibody production. Adv. Drug Deliver Rev. 2006, 58, 671–685. [Google Scholar] [CrossRef]
- Shukla, A.A.; Hubbard, B.; Tressel, T.; Guhan, S.; Low, D. Downstream processing of monoclonal antibodies—Application of platform approaches. J. Chromatogr. B 2007, 848, 28–39. [Google Scholar] [CrossRef]
- Bhambure, R.; Kumar, K.; Rathore, A.S. High-throughput process development for biopharmaceutical drug substances. Trends Biotechnol. 2011, 29, 127–135. [Google Scholar] [CrossRef]
- Heckathorn, R.; Adams, D.; Hunter, J.; Frieden, E. Increasing Upstream Process Development Efficiency by Implementing Platform Glutamine Synthetase Cell Culture Processes. In Cells and Culture; Noll, T., Ed.; Springer Netherlands: Heidelberg, Germany, 2010; pp. 245–251. [Google Scholar]
- Del Val, I.J.; Kontoravdi, C.; Nagy, J.M. Towards the implementation of quality by design to the production of therapeutic monoclonal antibodies with desired glycosylation patterns. Biotechnol. Prog. 2010, 26, 1505–1527. [Google Scholar] [CrossRef]
- Gottschalk, U. Process Scale Purification of Antibodies: Downstream Processing of Monoclonal Antibodies: Current Practices and Future Opportunities, 1st ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Sommerfeld, S.; Strube, J. Challenges in biotechnology production—Generic processes and process optimization for monoclonal antibodies. Chem. Eng. Process. 2005, 44, 1123– 1137. [Google Scholar] [CrossRef]
- Shukla, A.A.; Hinckley, P. Host cell protein clearance during protein A chromatography: Development of an improved column wash step. Biotechnol. Prog. 2008, 24, 1115–1121. [Google Scholar] [CrossRef]
- Tarrant, R.D.R.; Velez-Suberbie, M.L.; Tait, A.S.; Smales, C.M.; Bracewell, D.G. Host cell protein adsorption characteristics during protein A chromatography. Biotechnol. Prog. 2012, 28, 1037–1044. [Google Scholar] [CrossRef]
- Royce, J. High-capacity protein A chromatography medium for mAb capture from hightiter feeds. BioProcess Int.2014, 12, 40–41. [Google Scholar]
- Lain, B.; Cacciuttolo, M.A.; Zarbis-Papastoitsis, G. Development of a high-capacity Mab capture step based on cation-exchange chromatography. BioProcess Int. 2009, 26–34. [Google Scholar]
- Lain, B. Protein A: The life of disruptive technology. BioProcess Int. 2013, 11, 29–38. [Google Scholar]
- Ghose, S.; Hubbard, B.; Cramer, S.M. Evaluation and comparison of alternatives to Protein A chromatography: Mimetic and hydrophobic charge induction chromatographic stationary phases. J. Chromatogr. A 2006, 1122, 144–152. [Google Scholar] [CrossRef]
- Konstantin Konstantinov, VP, Late Stage Process Development, BioRealization, Sanofi, (noted figures, via e-mail).
- ICH Q3, Q7, Q8, Q8(R2), Q9, Q10, and Q11
- FDA Guidance for Industry PAT A Framework for Innovative Pharmaceutical Development, Manufacturing and Quality Assurance
- Guideline on process validation for finished products - information and data to be provided in regulatory submissions, 21 November 2016, EMA/CHMP/CVMP/QWP/ BWP/70278/2012-Rev1,Corr.1
- EMA Guidelines on Process validation, 29 March 2012, EMA/CHMP/CVMP/ QWP/70278/2012-Rev1
- Guideline on the use of near infrared spectroscopy by the pharmaceutical industry and the data requirements for new submissions and variations, 27 January 2014 EMEA/CHMP/ CVMP/QWP/17760/2009 Rev2
- Guideline on Real Time Release Testing (formerly Guideline on Parametric Release), 29 March 2012, EMA/CHMP/QWP/811210/2009-Rev1