Introduction
The cryopreservation process can be stressful to sensitive microorganisms, including clinical isolates and stressed, injured or damaged environmental isolates. Conventionally, microorganisms are harvested in late logarithmic to early stationary phase prior to cryopreservation and are subsequently stored at -80°C.1-3,6 The timing of cell harvest has been reported to be critical and may affect the survival rate of bacterial cells after freezing.2 This traditional method, however, may not be ideal for all microorganisms. Sensitive microorganisms may benefit from more careful cryopreservation which uses cell growth data based on viable cell counts in addition to OD600, as well as the implementation of controlled rate freezers and storage in the vapor phase of liquid nitrogen. The overall health of the microbial culture at harvest, the cryopreservative medium, the cryoprotective agent and the cryopreservation method can all have significant impacts on the recovery of cryopreserved cells.
During cryopreservation, ice crystal formation can occur outside of cells when the temperature declines slowly. As a result, water migrates out of cells and the intracellular solute concentration increases. However, when a rapid cooling takes place, cells are able to retain more water intracellularly and ice crystals form within the cell.2,3 Controlling the rate of freezing is critical in order to keep the amount of solute and water balanced intracellularly and to prevent the formation of ice crystals in the interstitial cell wall. While passive freezing is used routinely, fastidious organisms, clinical isolates, stressed, damaged, or injured environmental isolates may require better control throughout the freezing process. In this regard, controlled rate freezing is superior in that a desired temperature-control paradigm can be easily realized. Typically, during controlled rate freezing, the temperature initially ramps down at a rate of 1°C to 3°C per minute until reaching -40°C, followed by a second-phase decrease at a rate of 10°C per minute to -90°C (or the final temperature for storage).2,6
For long-term preservation of microorganisms, it is generally recommended to store cell banks in liquid nitrogen (below -196°C) or in the vapor phase of liquid nitrogen (-130°C to -190°C). Cryopreserved microbial cells kept at ultra-low temperatures are thought to retain their viability and genetic stability.4 In addition, some studies showed that -70°C was almost as effective as liquid nitrogen for long-term storage.4-6 In comparison, other studies suggested that storing cell banks at -20°C is not ideal for long-term stability.6
Historically at Pfizer, optical density was predominantly used to determine the growth phase of microbial cell cultures. Microorganisms were typically harvested between late logarithmic and early stationary phases at an OD600 of no less than 2.67, a value set by our discovery partners. However, there was little information with regard to cell health and viable cell counts, which is now incorporated along with optical density to determine the growth phase. Our recent investigation showed that for some microorganisms, cell viabilities began to decline even before the OD600 requirements were met, resulting in the cryopreservation of unhealthy cells. This observation in turn led us to embark on the improvement of the cryopreservation practice, which is expected to provide optimal conditions for microbial cell banking and to maintain high cell viability after long-term storage. In this report, we compare the historical and the current methods for microbial cryopreservation with an emphasis on cell bank stability.
Methods
For the historical method of cultivating and cryopreserving microorganisms, the decision of harvest solely relies on whether a target optical density is met regardless of the growth phase and cell health. After the harvest, cells were cryopreserved and frozen in -80°C freezers. In comparison, the current method uses viable cell counts and controlled-rate freezers for optimization (Figure 1).
 Figure 1. Cryopreservation process comparison
Figure 1. Cryopreservation process comparisonTo compare these methods, vials from two clinical isolates of the Streptococcaceae family were thawed, cultivated, harvested and cryopreserved using both historical and current methods. OD600 ≥ 2.67 was used as the harvest criteria for the historical method and harvested cells were cryopreserved by the addition of 20% (v/v) glycerol and placement in a polycarbonate box stored directly into a -80°C freezer. For the current method, in order to determine the optimal point of harvest, a growth curve consisting of viable cell counts (CFU/mL) at different time points was developed in addition to the one consisting of OD600 (Figure 2). Viable cell counts were determined using the standard agar plate count method, which has been well documented in previous studies.7,8 The acceptable range of enumeration was between 25 and 250 colonies per plate and two scientists performed the viable cell count independently to minimize variability. Newly cultivated cells were harvested in the bona fide logarithmic growth phase (Figure 2, green circle), followed by cryopreservation by 20% (v/v) glycerol addition and freezing in a controlled-rate freezer. Once frozen, cryopreserved vials from both methods were randomly selected and stored at -20°C, -80°C or in the vapor phase of liquid nitrogen.

Figure 2. Growth curves consisting of either viable cell counts (CFU/ mL) or OD
600 readings. The green circle represents an optimal point of harvest where OD
600 and CFU/mL are both ascending, indicating a healthy culture in logarithmic growth phase.
To evaluate the stability of these cell banks, vials were recovered from each storage condition for viable cell counts at approximately one month intervals for the first six months and then every six months thereafter.
Results
A viable cell count is critical in assessing the health of the cell culture prior to cryopreservation, which may impact the stability of cryopreserved cultures during long-term storage. Therefore, it was used as the key measure to compare two methods of cryopreserving microbial cell cultures in this study.
As illustrated in Figure 3, Streptococcaceae A was harvested and cryopreserved when cell cultures reached an OD600 of 3.9 for the historical method and 2.3 for the current method. The higher OD600 observed for the historical method met the requirement of ≥ 2.67 and the lower OD600 observed for the current method met the requirement of the culture being in logarithmic phase. Despite cultures having different OD600, the viable cell counts were at the same order of magnitude (2.1 x 109 CFU/mL for the historical method vs. 1.5 x 109 CFU/mL for the current method) (Figure 4). In comparison, Streptococcaceae B was harvested and cryopreserved when cell cultures reached an OD600 of 3.3 for the historical method and 1.8 for the current method (Figure 3). The higher OD600 observed for the historical method met the requirement of ≥ 2.67 and the lower OD600 observed for the current method met the requirement of the culture being in logarithmic phase. However, approximately one log difference in viable cell counts was observed (2.5 x 108 CFU/mL for the historical method vs. 1.1 x 109 CFU/mL for the current method), indicating that unhealthy/dead cells were most likely cryopreserved using the historical method (Figure 4).
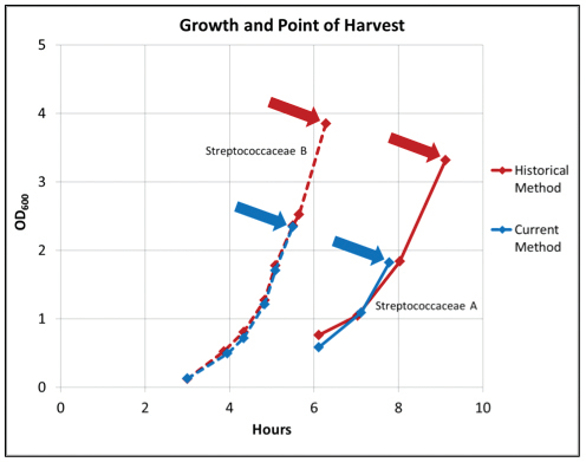
Figure 2. Growth curves consisting of either viable cell counts (CFU/ mL) or OD
600 readings. The green circle represents an optimal point of harvest where OD
600 and CFU/mL are both ascending, indicating a healthy culture in logarithmic growth phase.
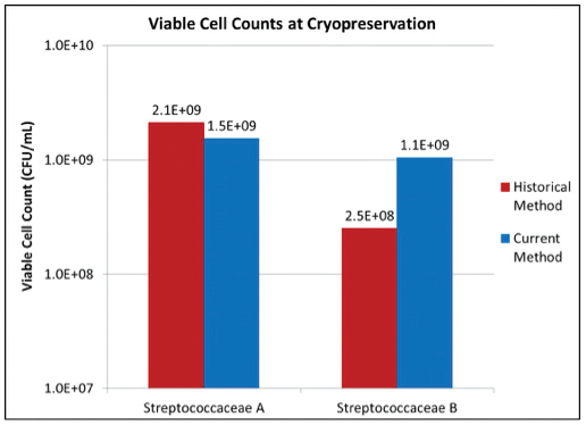 Figure 4. Viable cell counts determined by the standard plating method at cryopreservation for both Streptococcaceae organisms.
Figure 4. Viable cell counts determined by the standard plating method at cryopreservation for both Streptococcaceae organisms.We further evaluated the stability of cryopreserved cell cultures of both organisms at respective storage conditions. No decline in viable cell count was observed for either organism cryopreserved by either method when stored at -80°C or in the vapor phase of liquid nitrogen over 32 months (data not shown).
Interestingly, for both organisms, cryopreserved cells using the historical method that were subsequently stored at -20°C showed a more rapid decline in viable cell counts in our stability study. Streptococcaceae A started with a viable cell count of 2.1 x 109 CFU/mL and declined to 33 CFU/mL, an approximate 8-log reduction after 32 months (Figure 5). Streptococcaceae B started with a viable cell count of 2.5 x 108 CFU/mL and declined to 2.1 x 104 CFU/mL, an approximate 4-log reduction after 32 months in storage (Figure 5).
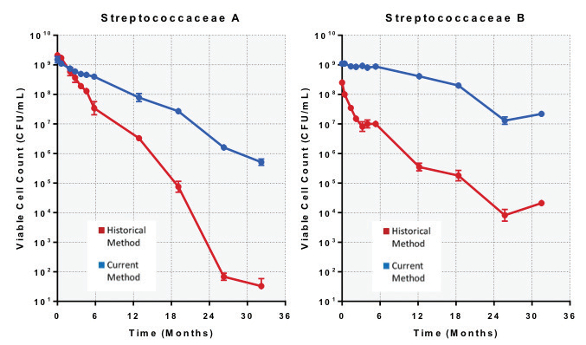 Figure 5. Stability study over 32 months showing viable cell counts for cryopreserved Streptococcaceae organisms stored at -20°C.
Figure 5. Stability study over 32 months showing viable cell counts for cryopreserved Streptococcaceae organisms stored at -20°C.A slower rate of decline was observed for both organisms cryopreserved by the current method and subsequently stored at -20°C. Streptococcaceae A started with a viable cell count of 1.5 x 109 CFU/mL and declined to 5.1 x 105 CFU/mL, an approximate 3.5- log reduction after 32 months (Figure 5). Streptococcaceae B started with a viable cell count of 1.1 x 109 CFU/mL and declined to 2.2 x 107 CFU/mL, an approximate 2-log reduction after 32 months in storage (Figure 5).
Conclusion
It is known that -20°C is not an optimal temperature for the long-term storage of microbial cell banks. However, such a sub-optimal condition can be used in a microbial cell bank stability study to demonstrate accelerated cell culture degradation over a reasonable time frame of storage (< 3 years in our study), as cryopreserved cells stored at -80°C or in the vapor phase of liquid nitrogen may not show any sign of degradation in a short-term study, as was the case in this work. The results showed that at a storage temperature of -20°C, the viable cell counts of both Streptococcaceae organisms cryopreserved using the current method had a slower decline. This observation indicates that the current method is more robust than the historical method and it is expected that the current cryopreservation method is able to provide greater long-term stability of microbial cell banks than the historical method. Our results demonstrate the principle of using a sub-optimal storage temperature to generate an accelerated degradation in a short time frame.
References
- Chang, L.T. and Elander, R.P. 1986. Long Term Preservation of Industrially Important Microorganisms. Manual of Industrial Microbiology and Biotechnology, Ed. Demain, A.L. and Solomon, N.A. American Society for Microbiology. 49-55.
- Gherna, R.L. and Flickinger, M.C. 2009. Culture Preservation. Encyclopedia of Industrial Biotechnology. John Wiley & Sons, Inc. 153-161.
- Simione, F.P. and Thermo Fisher Scientific Inc. 2009. Thermo Scientific Nalgene and Nunc Cryopreservation Guide. 2-5. https://www.atcc.org/~/media/PDFs/Cryopreservation_ Technical_Manual.ashx
- Perry, S.F. 1995. Freeze-Drying and Cryopreservation of Bacteria. Methods Mol Biol. 38:21-30.
- Heckly, R.J. 1978. Preservation of Microorganisms. Advances in Applied Microbiology. Ed. Perlman, D. Academic Press. 24:1-53.
- Tedeschi, R and De Paoli, P. 2011. Collection and Preservation of Frozen Microorganisms. Methods Mol Biol. 675:313-26.
- Albus, W.R. 1928. Some Observations on the Plate-Count Method of Enumerating Bacteria in Milk. J Bacteriol. 16:269-277.
- Sutton, S. 2012. The Limitations of CFU: Compliance to CGMP Requires Good Science. Journal of GXP Compliance. 16:74-80.