Bioavailability enhancing (BAE) technologies for oral drug delivery may be designed to overcome many different hurdles to absorption. The critical aspect(s) limiting systemic exposure may include one or multiple rate-limiting steps that formulation may address. These may include (1) solubility, (2) dissolution rate; (3) precipitation of drug in the gastrointestinal (GI) tract; (4) diffusion of drug through the aqueous boundary layer (ABL); (5) permeation across epithelial cells; (6) efflux; (7) API chemical degradation in the GI tract; (8) and drug-drug interactions. The specific rate limiting steps for absorption depend on both the drug product and the individual drug substance.
Several classification systems have been proposed to aid in binning compounds for guiding both formulation and testing methodology. The most commonly referenced system is the biopharmaceutics classification system (BCS), dividing compounds based on solubility and permeability. The BCS II and IV classes have been further divided into subclasses of acidic, basic, and neutral compounds to aid in the selection of in vitro methodology and parameters.1 Other classification systems are also useful for determining the rate limiting step(s) to absorption.
The fraction absorbed classification system (FaCS)2 distinguishes cases where absorption is limited by dissolution rate, permeability, or the combination of solubility and permeability. Permeability limitations are further categorized as being limited across the ABL or the epithelial membrane. These classifications have been applied to drug substances as well as drug product properties as a way to predict food effects, identify enabling technologies, and determine bioequivalence strategies.
Formulators have many technologies to select from in order to solve BAE challenges. Selecting an effective technology requires matching the problem statement for the active pharmaceutical ingredient (API) with an appropriate formulation strategy. The problem statement is described by the hurdles to absorption listed above together with the required dosage form, chemical stability, cost, and other issues defining the target product profile. Common BAE technologies include salt forms, micronization, amorphous formulations, cocrystals, and lipid based formulations.
Once an appropriate strategy has been identified, it is important to test formulations using in vitro methods that indicate performance related to the in vivo rate limiting step(s) to absorption and/or likely failure mechanisms of the formulations. This approach will maximize the likelihood of developing a successful formulation.
The first step in designing a biorelevant dissolution test is to determine the problem statement to be addressed. For BCS II and IV drug products, the most common problem statements can be categorized as relating to precipitation, dissolution rate, or solubilitypermeability limited absorption. Within each of these categories, there are several sub-classes. A simplified strategy for selecting a dissolution test is shown in Figure 1. In reality, the design process is more complex – each API and formulation is unique and a dissolution test should be designed to capture its individual critical performance attributes. The goal of a formulator should be to select the simplest dissolution test that predicts relative in vivo performance of the formulations being assessed.
 Figure 1. Problem statement driven selection of in vitro tests for BCS II and IV drug products
Figure 1. Problem statement driven selection of in vitro tests for BCS II and IV drug productsMany BAE formulations are designed to lead to supersaturation in GI fluids. While supersaturation can lead to improved absorption, it also leads to the possibility of precipitation. A simple non-sink dissolution test can often determine if there is a potential for an API to precipitate on a biologically relevant timescale. If there is a potential for precipitation, the mechanism and critical factors affecting precipitation inform the type and complexity of the dissolution test to use.
Depending on the API and excipients utilized, precipitation may occur in either the stomach or the intestine. Additionally, intestinal precipitation may be dependent on exposure to gastric fluid with sensitivity to the pH of the gastric fluid and the exposure time. In these cases, a non-sink transfer dissolution test should be utilized. The simplest transfer test involves first adding gastric media to the formulation and mixing for a set period of time. Subsequently, a concentrated “intestinal buffer” is added to raise the pH of the final mixture to a physiological intestinal pH (5 – 7) and the bile salt concentration to a desired level. These two-stage dissolution tests are simple and can be performed in a USP dissolution apparatus, but do not capture physiologically relevant transfer of material from the stomach to the intestine.
A more involved option for transfer tests is a multi-compartment or controlled transfer dissolution test. The artificial stomach and duodenum (ASD),3 gastrointestinal simulator (GIS),4 and others involve first adding the formulation to a “gastric compartment” and then the apparatus transfers fluids and solids at a controlled feed rate from the gastric compartment to a “duodenal compartment.” Many of these tests include gastric and bile secretion to mimic physiological conditions along the GI tract. In Figure 1, all of these tests are termed “ASD.” These tests typically provide more insight into precipitation mechanisms than a two-stage transfer test.
In both the two-stage and controlled transfer tests, the tendency to crystallize is a “worst case scenario” because API is not being removing by permeation as it would in vitro. When permeation is slow these tests can be discriminating for resistance to precipitation and sustainment of permeable API. When permeation is known to be rapid, however, it may be appropriate to use a dissolution test with a permeation step.
Dissolution tests with a permeation step include biphasic dissolution tests where the simulated GI media is in contact with a waterinsoluble organic phase,5 membrane flux tests incorporating a lipid-filled membrane (membrane is impermeable to micelles, inclusion complexes, etc.),6 and dissolution/permeation systems that incorporate Caco-2 or other cell monolayers to separate the dissolution and permeation compartments.7 By using a dissolution test with a permeation step, API is constantly removed from the system and the degree of supersaturation may be lowered, reducing the tendency to crystallize.
Lipid formulations contain digestible excipients (i.e. triglycerides and diglycerides). Such formulations should therefore be evaluated using dissolution tests that include enzymatic digestion of the lipidic components to assess the effect of digestion on precipitation of supersaturated drug. The digestion products of lipid formulations often have a large impact on the degree of drug solubilization and therefore precipitation and absorption.8
When dissolution is the rate limiting step to absorption, the impact of formulation and process parameters on dissolution rate is key to an in vitro dissolution testing strategy. The simplest method for assessing dissolution rate is a sink dissolution test. Sink dissolution tests are performed such that the rate of dissolution is insensitive to the concentration of dissolved API in the media. Sink conditions are often not biorelevant for BCS II or IV compounds, but the data are valuable as input to mechanistic modeling software. For example, the impact of dissolution rate on absorption can be assessed by modelling dissolution in conjunction with in vivo processes such as gastric emptying, intestinal transit, GI hydrodynamics, and absorption rate.
When dissolution rate in the duodenum is critical and it is desirable to compare dissolution rate relative to gastric emptying time, an ASD-like test can be utilized. In such a test, it is clear when dissolution is slower than gastric emptying and the impact on intestinal concentration can be measured. Another option for comparing the dissolution rate of formulations after gastric exposure, is to perform a two-stage test where the second stage of the dissolution test is a sink condition.
An alternative method for assessing dissolution rate is the use of a dissolution/permeation test. The key parameters for designing a permeation test that is sensitive to dissolution rate have been discussed in detail. One advantage of this approach is when dissolution rate changes over time in intestinal media due to aggregation, Ostwald ripening, or use of controlled release formulations. With a dissolution/ permeation test, these changes can be monitored over physiological time scales.9
For solubility-permeability limited absorption, dissolution tests with a permeation step are ideally suited for formulation assessment. Depending on the API, the formulation, and the test setup, permeation may be limited by either ABL diffusion or diffusion across the membrane or cell monolayer. This type of test is an excellent choice for formulation assessment when designed to match the rate limiting step of the test with the rate limiting step known (or predicted) in vivo. When membrane diffusion is limiting, drug activity determines the flux of drug across the membrane. When ABL diffusion is limiting, all rapidly diffusing drug species (e.g. neutral drug, charged drug, micelle-bound drug, and nanoparticles) may impact flux.
By way of example, itraconazole (ITZ) is a very low solubility, highly permeable (BCS II) antifungal which is available commercially as an amorphous spray layered dispersion, Sporanox®. In a recent publication, we explored SDDs of itraconazole that form varying concentrations of drug-rich colloids (ca. 150 nm) and compared their performance to Sporanox® both in vitro and in vivo. 10 ITZ is solubility- ABL permeability limited (SL -U) according to the FaCS for both the crystalline and amorphous forms of the drug (Table 1). For SL-U compounds, formulations which increase the total solubilized drug (through formation of micelles, colloids, nanoparticles, etc.) are predicted to improve oral bioavailability by increasing diffusion across the ABL.
Table 1. Properties and classification of crystalline and amorphous itraconazole (200 mg dose).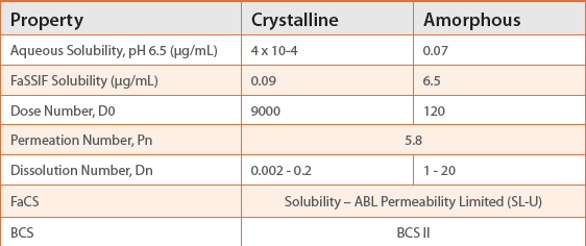
Following the guidance of Figure 1, a dissolution/permeation assay was used to compare spray dried dispersions (SDDs) of ITZ. All three SDD formulations produced drug-rich colloids and the permeation rate in the in vitro assay increased with an increasing concentration of drug-rich colloids. In order to predict the performance in an in vivo rat study, a pH 6.5 buffer with a high concentration (27 mM) of bile salt was selected as the donor media. The acceptor phase media (2% SLS) was selected to provide much higher solubility than the donor media and the two compartments were separated by a lipid-filled membrane. The apparent concentration of ITZ reached for the SDDs was 2 – 20x higher than that of the control formulation. The resulting flux was 1.5 – 2.2x higher than for the control formulation (Figure 2).
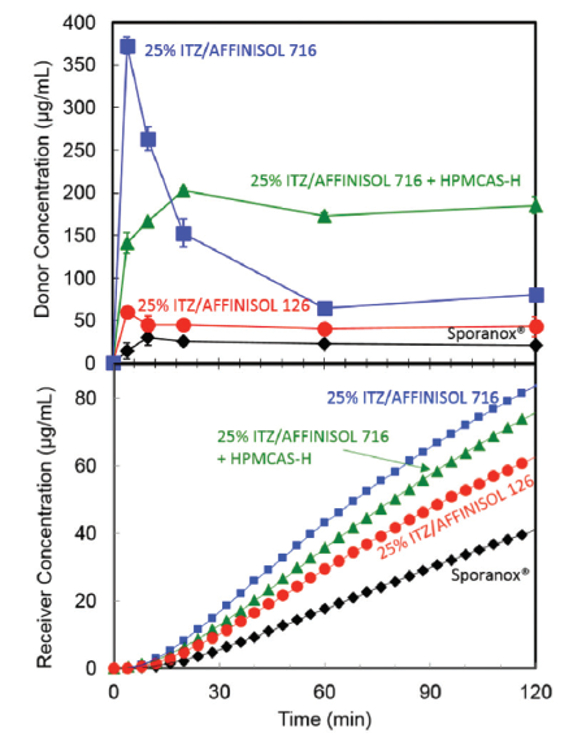 Figure 2. Donor (top) and receiver (bottom) concentration of itraconazole for 3 SDD formulations and the commercial spray layered dispersion, Sporanox® in a dissolution/ permeation experiment.
Figure 2. Donor (top) and receiver (bottom) concentration of itraconazole for 3 SDD formulations and the commercial spray layered dispersion, Sporanox® in a dissolution/ permeation experiment.The measured in vitro flux increased with increasing concentration of drug-rich colloids. The in vitro flux was an excellent predictor of relative absorption rate in rats (Figure 3).
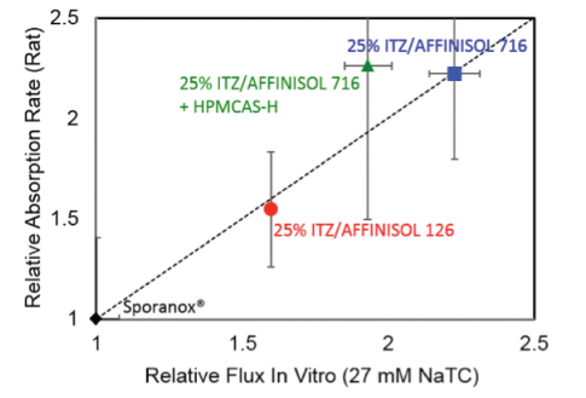 Figure 3. Relative In vivo vs. in vitro absorption rate of three SDD formulations relative to the commercial Sporanox® formulation
Figure 3. Relative In vivo vs. in vitro absorption rate of three SDD formulations relative to the commercial Sporanox® formulationThis example demonstrates one case where careful selection of the in vitro test and the conditions of the test allows for in vivo prediction beyond simple rank ordering of formulations. Utilizing relatively simple in vivo predictive dissolution tests early in development can aid in selecting an optimal formulation and balancing the bioperformance of formulations with other critical attributes of the drug product such as stability and manufacturability.
Selection of a proper in vitro test must be based on an understanding of the hurdles to in vivo absorption. In fast-paced formulation development labs it can be tempting to develop “one size fits all” tests for formulations. Such an approach, however, can be misleading in an environment where multiple compounds each with unique properties are under development. By following the guidance outlined here, it is possible to test formulations using in vivo predictive dissolution tests in a streamlined fashion while individually addressing the variety of problem statements presented by a diverse portfolio of compounds.
References
- Tsume, Y., Mudie, D. M., Langguth, P., Amidon, G. E. & Amidon, G. L. The Biopharmaceutics Classification System: Subclasses for in vivo predictive dissolution (IPD) methodology and IVIVC. Eur. J. Pharm. Sci. 57, 152–163 (2014).
- Sugano, K. & Terada, K. Rate- and Extent-Limiting Factors of Oral Drug Absorption: Theory and Applications. J. Pharm. Sci. 104, 2777–2788 (2015).
- Polster, C. S., Atassi, F., Wu, S. J. & Sperry, D. C. Use of artificial stomach-duodenum model for investigation of dosing fluid effect on clinical trial variability. Mol. Pharm. 7, 1533– 1538 (2010).
- Matsui, K., Tsume, Y., Takeuchi, S., Searls, A. & Amidon, G. L. Utilization of gastrointestinal simulator, an in vivo predictive dissolution methodology, coupled with computational approach to forecast oral absorption of dipyridamole. Mol. Pharm. 14, 1181–1189 (2017).
- Shi, Y. et al. Assessing Supersaturation and Its Impact on In Vivo Bioavailability of a LowSolubility Compound ABT-072 With a Dual pH, Two-Phase Dissolution Method. J. Pharm. Sci. 105, 2886–2895 (2016).
- Stewart, A. M. et al. Development of a Biorelevant, Material-Sparing Membrane Flux Test for Rapid Screening of Bioavailability-Enhancing Drug Product Formulations. Mol. Pharm. 14, 2032–2046 (2017).
- Kataoka, M. et al. Application of Dissolution/Permeation System for Evaluation of Formulation Effect on Oral Absorption of Poorly Water-Soluble Drugs in Drug Development. Pharm. Res. 29, 1485–1494 (2012).
- Williams, H. D. et al. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 3: Understanding supersaturation versus precipitation potential during the in vitro digestion of type I, II, IIIA, IIIB and IV lipid-based formulations. Pharm. Res. 30, 3059–3076 (2013).
- Mudie, D. M. et al. Mechanistic analysis of solute transport in an in vitro physiological twophase dissolution apparatus. Biopharm. Drug Dispos. 32, 378–402 (2012).
- Stewart, A. M. et al. Impact of Drug-Rich Colloids of Itraconazole and HPMCAS on Membrane Flux in Vitro and Oral Bioavailability in Rats. Mol. Pharm. 14, 2437–2449 (2017).
Author Biography
Michael Grass is a Senior Research Chemist at Lonza Pharma & Biotech where he develops methods, conducts testing, and analyzes data for a wide range of pharmaceutical formulations. His expertise includes formulation development of challenging compounds, dissolution testing, and physical stability of amorphous dispersions.