Introduction
Even with an ever-increasing array of cutting edge technologies at its disposal, the process of drug discovery continues to be a difficult one. As has been well documented, increased investments in pharmaceutical research and development have not led to a concomitant rise in the number of new drugs.1 This certainly has not been for a lack of new targets, as seemingly every week results of genomic and proteomic studies report the association of one or more genes or proteins to a disease state. These significant advances, however, have not been followed by a concurrent increase in the availability of new chemical matter against which to interrogate this myriad of targets. In the 1990s, combinatorial chemistry was hailed as a revolutionary approach that would permit a meteoric advance in the identification of novel therapeutic molecules.2 Although successful to some degree with associated techniques, such as parallel synthesis, solid phase methodology, high-throughput compound profiling and purification, now entrenched into modern drug discovery efforts,3 the perceived failure of this technology to deliver on its surrounding hype and the subsequent unrealistic expectations built thereupon, resulted in a hesitancy by many in the industry to rush into the adoption of new chemical technologies.
Further, associated with the resurgence of novel targets, a dramatic shift in drug discovery approaches to biological entities has contributed to a greater number of antibody and protein drug products appearing and advancing in the pipelines of pharmaceutical and biotech companies. Considering the therapeutic success, faster approval, lower clinical failure rate, and premium pricing being realized by these biomolecules, the attraction is certainly alluring.4 Hence, while advances such as CRISPR/Cas9 and CAR-T capture the headlines and imagination of investors and scientists, chemical technologies have often been forgotten. However, biologics do possess some significant limitations. Importantly, they can only address cell surface targets, since they are too large to cross cellular membranes. In addition, their preparation is complex, expensive and difficult to scale, plus concerns about soliciting an undesirable immune response are ever-present.5 Indeed, despite the growing impact of biologics, small molecule chemical drugs still account for the lion’s share of global sales.
Unfortunately, attempts to modulate intracellular pathways and targets that are inaccessible to biologics have also proven intractable to the traditional compounds that dominate historical corporate compound collections. The limitations of biological molecules coupled with the difficulties encountered with small molecules has led to consideration of non-traditional structures for modulation of these challenging targets, such as protein-protein interactions (PPI), protein-nucleic acid interactions and transcription factors.6
One particularly attractive chemical class that suits this purpose has been macrocyclic compounds.7 Macrocycles are molecular structures that contain one or more rings of at least 12 atoms with the unique characteristic that they combine the benefits of large biomolecules, such as high potency and exquisite selectivity, with those of small molecules, including reasonable manufacturing costs, favorable pharmacokinetic properties, including oral bioavailability, ease of administration and lack of immunogenicity. Interest in these structures originated in part from the many macrocyclic natural products that have proven utility as therapeutic agents (e.g. vancomycin, amphotericin B, romidepsin, sirolimus). Further, the pre-organized and semi-rigid character of these structures holds potential for excellent activity, even against intractable targets. Indeed, these molecules possess dense functionality packed into a compact, accessible scaffold, yet still maintain sufficient flexibility to enable effective binding at disparate sites. In addition, macrocycles possess defined topologies, which position functional groups in specific regions of space for target interaction, thereby permitting high potency to be achieved, even early in a discovery program. In particular, they are capable of effective interactions at the large, shallow surfaces that characterize many of the challenging target classes.
Macrocycles: Technology Development
Despite their favorable nature, however, not much excitement surrounded the area of macrocyclic drug discovery when the author entered the field in 1999. Indeed, there was actually considerable resistance to the acceptance of macrocycles as possible pharmaceutical agents since they did not fit the usual structural paradigm that had served the industry very well for decades – small molecular weight (MW) heterocyclic molecules with a limited number of functional groups that, more often than not, conformed to what became known as Lipinski’s rule of five8 or, more succinctly termed, “drug-likeness.” Further, synthetic accessibility for macrocyclic structures has been a long-standing challenge, particularly in the more diverse library format needed for the high throughput screening (HTS) efforts central to the majority of modern drug discovery programs.
 Figure 1. Macrocycle Chemical Space7b
Figure 1. Macrocycle Chemical Space7bAs with many nascent fields, it fell to smaller entrepreneurial firms to change this situation and alter the perception of macrocycles in the pharmaceutical industry. In 2000, only two companies were actively pursuing macrocyclic structures as a core technology: Polyphor in Switzerland, and the author’s former company, NéoKimia (later Tranzyme Pharma9 ) in Canada. Their proprietary technologies proved successful in advancing macrocycles into the clinic: the antibiotic murepavadin (POL7080)10 and the gastrointestinal prokinetic agents ulimorelin11 and TZP-10212 (Figure 2).
 Figure 2. Initial Macrocyclic Clinical Candidates
Figure 2. Initial Macrocyclic Clinical CandidatesRoused by these successes, a realization gradually developed in the industry that there actually were biologically interesting molecules in this “beyond rule of five” (bRo5) space.6 Hence, the situation steadily began to change from that humble beginning and, during the first decade of the twenty-first century, an array of innovative technologies were created that provided access to macrocyclic compounds of varying size and composition. These approaches ranged from purely chemical in nature13 to hybrid strategies that combined biological methods, such as phage or mRNA display, intein-mediated protein splicing, engineered enzymes and directed in vitro translation systems, with one or more chemical steps.14 The latter methods, in particular, can generate very large numbers of compounds, but such macrocycles also tend to be highly peptidic and in the higher MW range, bringing concerns regarding their ability to possess drug-like properties. In contrast, technologies that rely solely on chemical transformations generally produce compounds occupying the lower MW end of the macrocycle continuum with their concomitant improved potential for favorable physicochemical and pharmacokinetic (PKADME) profiles.
Today, the landscape is dramatically different with approximately thirty companies embracing macrocycle-based technologies as a core asset within their drug discovery effort, and many others employing them for selected projects (Table 1). Indeed, a sure sign this area has moved into the “established” phase is that multiple vendors are now offering macrocyclic libraries for sale. The temptation then might be to consider macrocycles as now becoming just another commodity, although that also would not be appropriate, since a vast region of macrocycle chemical space still remains unexplored with current compounds and certain key deficiencies in working with these structures still exist. Indeed, molecular modeling and PKADME properties are two important aspects where significant challenges remain and current research efforts are being directed.
Table 1. Selected Macrocycle Technology Companies and Library Vendors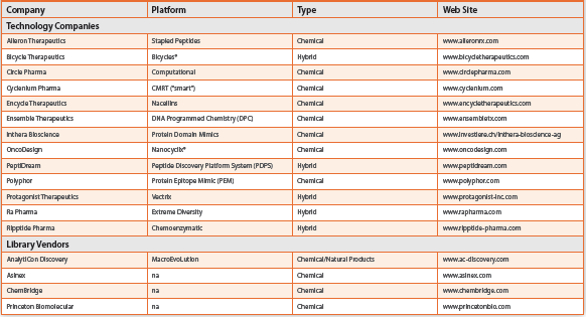
However, initial concerns about the ability to scale-up macrocycles, which the author recalls being emphatically told at one point would preclude such molecules from ever becoming viable therapeutics, were able to be decisively surmounted by the results with the first clinical HCV macrocyclic protease inhibitor, ciluprevir (BILN 2061), when a synthesis that was successful in preparing over 400 kg of the active ingredient was reported.15

Macrocycles: Challenges
As mentioned earlier, a host of hybrid chemico-biological technologies have resulted in the production of very large numbers of generally high MW macrocyclic compounds. Such macrocycles have proven themselves very effective at modulating a range of pharmacologically relevant interactions and providing insight into the structural determinants thereof.14 However, these large macrocycles typically possess considerable peptidic character, which, in concert with their size, create significant issues in attaining the necessary pharmacokinetic profile required for development as therapeutic agents. Indeed, it is likely that they fail to satisfy any of the iconic rule of five criteria.8 Even smaller MW macrocycles with high amide bond content can often encounter these same problems. For that reason, alternative guidance (“rules”) specifically for macrocyclic molecules and libraries has been defined employing some of the same measures that were used to formulate rule of five criteria, as well as other parameters more appropriate for this structural class.16 Compared to small molecules, where relevant data on large collections of diverse compounds are available, such an undertaking is limited by the number of macrocycles for which sufficient information is available. Hence, this analysis is skewed by the sizeable quantity of natural product structures included, since they have, to a large extent, been the most thoroughly studied compounds. As an example, for a compilation of 125 orally absorbed cyclic peptides, which violated the rule of five limits to different degrees, it was found that oral bioavailability was generally favored by a lower number of hydrogen bond donors and reduced flexibility.17 Nonetheless, careful investigations of cyclic natural products have provided insight into factors that are critical to obtaining favorable properties in macrocyclic compounds with an emphasis on passive cellular permeability as a surrogate marker for oral bioavailability.
In particular, cyclosporine is an 11-amino acid peptide with potent immunosuppressive effects18 that is often presented as the poster child for macrocyclic peptide drugs. However, it is actually the only one of the 30 approved macrocyclic peptide drugs that is dosed orally. This has been explained by the chameleonic character of the macrocycle in being able to adapt its structure to the local environment.19 Cyclization has been well-known to render peptidic molecules more resistant to degradation by shielding vulnerable functionalities from metabolic enzymes. In an analogous fashion, the cyclosporine structure exposes its polar functionalities when in an aqueous medium, but adopts a folded, almost micellular, structure with the polar groups shielded on the interior through formation of a network of intramolecular hydrogen bonds to enable traversal of the lipid membrane (Figure 3).
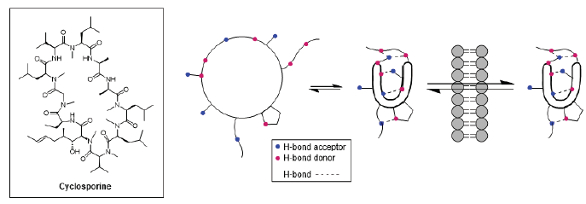 Figure 3. Chameleonic Behavior of Macrocycles19
Figure 3. Chameleonic Behavior of Macrocycles19That this ability is not unique to cyclosporine was shown by a study of 39 cyclic peptide natural products, where it was found that good passive cell permeability was exhibited by those that had low energy conformations in which compounds could effectively hide polar functionalities using tactics such as by N-methylation, bulky side chains and intramolecular hydrogen bonds (IHB).20 Additionally, in an evaluation of cyclic peptide libraries containing only N-methyl residues to remove the effects of IHB on permeability, considerably reduced permeability was observed at MW > 1000 Da, suggesting that this could represent an upper size limit for these compounds with respect to oral availability.21
Although these elegant studies have advanced the understanding of the factors involved in imparting desired properties into a peptidic structural framework, the applicability to non-peptidic macrocyclic molecules was less clear. Recently, however, a detailed investigation using 200 such macrocycles from a designed collection demonstrated that the key to their favorable drug-like properties also resided in their capacity to modify their conformation to match the polarity of the local environment.22 These studies revealed that stereo- and regiochemistry also have a significant influence on some of these properties, including cellular permeability.
Of course, as with traditional medicinal chemistry optimization, modifications made specifically to benefit compound properties must always be balanced with maintaining the desired level of target activity and selectivity. In this respect, a recurring theme from these works is that the cyclic nature of these molecules is the proverbial doubleedged sword, with their rigid nature and defined conformations leading to high potency and selectivity. However, within a macrocycle, small structural modifications can result in significant conformational changes, so the balancing act between potency and other properties, such as PK-ADME, can prove to be even more delicate than with traditional small molecules. On the other hand, this characteristic also leads to optimization of desired properties with macrocycles not requiring major structural additions, hence does not significantly increase MW, in contrast to the situation with traditional small molecules.23 From this discussion, it can be concluded that the factors influencing the “drugability” of macrocyclic molecules are beginning to be understood, but the worth of this knowledge in enabling drug discovery still remains to be proven.
Another aspect of macrocycle drug discovery that is consistent with small molecule efforts is that structural biology in conjunction with molecular modeling has proven to have significant utility. In particular, such studies are beneficial for determining if cyclization of a structure might be an effective strategy for constraint into the bioactive conformation. Indeed, prior to the dedicated library efforts described herein, this was the primary manner in which synthetic macrocycles found their way into pharmaceutical discovery. However, in contrast to small molecules, the use of computational methods for de novo design and virtual screening in the macrocyclic area are complicated by the limited ability of current approaches to handle large ring systems, the accessibility of multiple low energy conformations, difficulties in pose prediction and imperfect treatment of conformational dynamics.24 Despite this, improvements are being made and many commercial programs now contain modules specifically aimed at handling conformational searches of macrocyclic molecules.25 However, it likely will still be some time before computer-assisted macrocycle design enters the discovery lexicon.
Macrocyclic Pharmaceuticals
Despite these continuing challenges, a growing number of synthetic macrocycles have progressed into the clinic with several already being established in the marketplace. An excellent compendium of macrocyclic compounds in clinical development and that have been approved for human treatment has been published.26 Importantly, their toxicological profiles have been found to more resemble those of traditional small molecules than biologics and, generally, have exhibited excellent safety. Although the impetus for the interest in macrocycles came from their potential for difficult targets, the many advantages provided by this compound class have made them quite valuable for the modulation of the more easily druggable target classes as well. In particular, macrocycles have impacted therapy for hepatitis C, where macrocyclic HCV NS3-4A protease inhibitors (Figure 4), grazoprevir (MK-5172), paritaprevir (ABT-450), simeprevir (TMC-435350), and vaniprevir (MK-7009), have helped to revolutionize the treatment of this disease and, in combination with other drugs, have led to this condition even being considered cured in infected individuals.27
 Figure 4. Marketed Macrocyclic Pharmaceuticals
Figure 4. Marketed Macrocyclic PharmaceuticalsAdvances in the construction of kinase inhibitors with unique properties have also been at the forefront of progress in the macrocycle area, with lorlatinib (Figure 5), an ALK/ROS1 tyrosine kinase inhibitor in Phase III clinical trials for metastatic non-small cell lung cancer,even demonstrating the ability to cross the blood-brain-barrier.28 As another example, pacritinib (Figure 5) is a dual JAK2 and FLT3 inhibitor that holds promise for the treatment of myelofibrosis and is also in Phase III.29
 Figure 5. Selected Clinical Macrocyclic Pharmaceuticals
Figure 5. Selected Clinical Macrocyclic PharmaceuticalsReturning to one of the primary drivers for the pursuit of macrocycles in drug discovery, the potential to address intractable targets such as PPI, the unique and versatile nature of the macrocyclic framework again has been demonstrated with increasing reports of successful modulation of this target class with these molecules, although such applications are still too early stage to gauge their clinical impact.30 Interestingly, in a comprehensive analysis of how drug molecules bind to their targets, it was found that macrocyclic drugs actually possess shapes that appear to be particularly appropriate for binding to the flat or shallow interaction surfaces characteristic of PPI.31
Lastly, one novel macrocycle class that deserves specific mention in the PPI area is “stapled” peptides. These molecules utilize side chain functional groups at particular residues that can react with each other to introduce a structural constraint (e.g. double or triple bond, triazole ring) into the peptide anchored at those residues, thereby stabilizing the secondary structure into a desired biologically relevant conformation, such as an α-helix, (Figure 6).32 An early example of this type of compound, ATSP-7041, which reactivates p53-mediated tumor suppression through dual inhibition of MDMX and MDM2, demonstrated efficacy in multiple human xenograft models.33 More recently, a later generation stapled peptide from this class, ALRN-6924, has entered clinical investigation for a variety of cancer indications.34 Applications for such molecules continue to be reported with the majority of them being in the modulation of PPI implicated in oncology.35
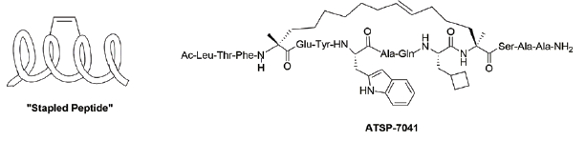 Figure 6. Examples of Stapled Peptides
Figure 6. Examples of Stapled PeptidesConclusion
From the discussion in this article, the impact of a new chemical class on drug discovery, even in these days of excitement over the latest biological breakthrough, cannot and should not be underestimated. Although the fervor surrounding the macrocycle area has subsided, careful, innovative and often elegant research from dedicated scientists in industry and academia over the past twenty years has led to the realization of the early promise of the macrocyclic compound class. Only a sampling of the advances and continuing challenges in this exciting area could be presented herein; the reader is directed to the articles cited in the references for additional information on the many inventive solutions that have been devised by the growing research community interested in macrocycles. Despite being intimately involved in this field for much of the past twenty years, the author is realistic enough to recognize that the progress in the macrocycle field falls short of being “revolutionary,” but its impact is nonetheless highly significant. The evolution of this area has proceeded very nicely, albeit not always smoothly, from early considerations related strongly towards technology development to now becoming established as another important element in the armamentarium of medicinal chemistry.
References
- Pammolli, F.; Magazzini, L.; Riccaboni, M. The productivity crisis in pharmaceutical R&D. Nat. Rev. Drug Discov. 2011, 10(6), 428-438.
- Gallop, M.A.; Barrett, R.W.; Dower, W.J.; Fodor, S.P.; Gordon, E.M. Applications of combinatorial technologies to drug discovery. 1. Background and peptide combinatorial libraries. J. Med. Chem. 1994, 37(9), 1233-1251; (b) Gordon, E.M. Barrett, R.W.; Dower, W.J.; Fodor, S.P.; Gallop, M.A. Applications of combinatorial technologies to drug discovery.Combinatorial organic synthesis, library screening strategies, and future directions. J. Med. Chem. 1994, 37(10), 1385-1401.
- Kennedy, P.; Williams, L.; Bridges, T.M.; Daniels, R.N.; Weaver, D.; Lindsley, C.W. Application of Combinatorial Chemistry Science on Modern Drug Discovery. J. Combi. Chem. 2008, 10(3), 345–354.
- (a) Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32(1), 40–51; (b) Smietana, K.; Siatkowski, M.; Møller, M. Trends in clinical success rates. Nat. Rev. Drug Disc. 2016, 15(6), 379–380.
- Lybecker, K.M. The Biologics Revolution in the Production of Drugs, Fraser Institute, July 2016.
- Indeed, this has been termed the “beyond rule of five” (bRo5) space, to differentiate from the space defined by compounds that fit the reference 8 criteria: Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21(9), 1115-1142.
- (a) Diggers, E.M.; Hale, S.P.; Lee, J.; Terrett, N.K. The exploration of macrocycles for drug discovery-an underexploited structural class. Nat. Rev. Drug Disc. 2008, 7(7), 608-624; (b) Terrett, N.K. Methods for the synthesis of macrocycle libraries for drug discovery. Drug Disc. Today: Technologies 2010, 7(2), e97-e104; (c) Marsault, E.; Peterson, M.L. Macrocycles are great cycles: applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J. Med. Chem. 2011, 54(7), 1961-2004; (d) Mallinson, J.; Collins, I. Macrocycles in new drug discovery. Fut. Med. Chem. 2012, 4(11), 1409-1438; (e) Yu, X.; Sun, D. Macrocyclic drugs and synthetic methodologies toward macrocycles. Molecules 2013, 18(6), 6230- 6268; (f) Yudin, A.K. Macrocycles: lessons from the distant past, recent developments, and future directions. Chem. Sci. 2015, 6(1), 30-49; (g) You, L.; An, R.; Liang, K.; Cui, B.; Wang, X. Macrocyclic Compounds: Emerging Opportunities for Current Drug Discovery. Curr. Pharm. Des. 2016, 22(26), 4086-4093.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23(1-3), 3-25.
- Now Ocera Therapeutics, which divested the MATCH™ macrocycle technology of Tranzyme to the Roche Group in December 2013.
- Luther, A.; Moehle, K.; Chevalier, E.; Dale, G.; Obrecht, D. Protein epitope mimetic macrocycles as biopharmaceuticals. Curr. Opin. Chem. Biol. 2017, 38, 45-51.
- Hoveyda, H.R.; Marsault, E.; Gagnon, R.; et al. Optimization of the potency and pharmacokinetic properties of a macrocyclic ghrelin receptor agonist (Part I): Development of ulimorelin (TZP-101) from hit to clinic. J. Med. Chem. 2011, 54(24), 8305−8320.
- Hoveyda, H.R.; Fraser, G.L.; Marsault, E.; et al. "Optimization of a macrocyclic ghrelin receptor agonist (Part II): Development of TZP-102." In E. Marsault, M.L. Peterson (Eds.) Practical Medicinal Chemistry with Macrocycles: Design, Synthesis, and Case Studies, Wiley, 2017, pp 545-558.
- Peterson, M. L. “The Synthesis of Macrocycles for Drug Discovery.” In J. Levin (Ed.) Macrocycles in Drug Discovery, Royal Society of Chemistry, Cambridge, 2014, pp 398-486.
- (a) Frost, J.R.; Smith, J.M.; Fasan, R. Design, synthesis, and diversification of ribosomally derived peptide macrocycles. Curr. Opin. Struct. Biol. 2013, 23(4), 571-580; (b) Bashiruddin, N.K.; Suga, H. Construction and screening of vast libraries of natural productlike macrocyclic peptides using in vitro display technologies. Curr. Opin. Chem. Biol. 2015, 24, 131–138.
- (a) Nicola, T.; Brenner, M.; Donsbach, K.; Kreye, P. First Scale-Up to Production Scale of a Ring Closing Metathesis Reaction Forming a 15-Membered Macrocycle as a Precursor of an Active Pharmaceutical Ingredient. Org. Proc. Res. Develop. 2005, 9(4), 513-515; (b) Ye. N.K.; Farina, V.; Houpis, I.N.; et al. Efficient large-scale synthesis of BILN 2061, a potent HCV protease inhibitor, by a convergent approach based on ring-closing metathesis. J. Org. Chem. 2006, 71(19), 7133-7145.
- Villar, E.A.; Beglov, D.; Chennamadhavuni, S.; Porco, J.A.,Jr.; Kozakov, D.; Vajda, S.; Whitty, A. How proteins bind macrocycles. Nat. Chem. Biol. 2014, 10(9), 723-731.
- Nielsen, D.S.; Shepherd, N.E.; Xu, W.; Lucke, A.J.; Stoermer, M.J.; Fairlie, D.P. Orally Absorbed Cyclic Peptides. Chem. Rev. 2017, 117(12), 8094-8128.
- Wenger, R.M.; Payne, T.G.; Schreier, M.H. Cyclosporine: Chemistry, Structure-Activity Relationships and Mode of Action. Prog. Clin. Biochem. Med. 1986, 3, 157-191.
- Whitty, A.; Zhong, M.; Viarengo, L.; Beglov, D.; Hall, D.R.; Vajda, S. Quantifying the chameleonic properties of macrocycles and other high-molecular-weight drugs. Drug Discov. Today 2016, 21(5), 712-717.
- Ahlbach, C.L.; Lexa, K.W.; Bockus, A.T.; Chen, V.; Crews, P.; Jacobson, M.P.; Lokey, R.S. Beyond cyclosporine A: conformation-dependent passive membrane permeabilities of cyclic peptide natural products. Fut. Med. Chem. 2015, 7(16), 2021-2030.
- Pye, C. R.; Hewitt, W. M.; Schwochert, J.; Haddad, T. D.; Townsend, C. E.; Etienne, L.; Lao, Y.; Limberakis, C.; Furukawa, A.; Mathiowetz, A. M.; Price, D. A.; Liras, S.; Lokey, R. S. Nonclassical size dependence of permeation defines bounds for passive absorption of large drug molecules. J. Med. Chem. 2017, 60(5), 1665−1672.
- Over, B.; Matsson, P.; Tyrchan, C.; Artursson, P.; et al. Structural and conformational determinants of macrocycle cell permeability. Nat. Chem. Biol. 2016, 12(12), 1065-1074.
- (a) Opera, T.I. Current trends in lead discovery: Are we looking for the appropriate properties? J. Computer-Aided Mol. Design 2002, 16, 325-334; (b) Wenlock, M.C.; Austin, R.P.; Barton, P.; Davis, A.M.; Leeson, P.D. A Comparison of Physiochemical Property Profiles of Development and Marketed Oral Drugs. J. Med. Chem. 2003, 46(7), 1250-1256.
- (a) Chen, I.J.; Foloppe, N. Tackling the conformational sampling of larger flexible compounds and macrocycles in pharmacology and drug discovery. Bioorg. Med. Chem. 2013, 21(24), 7898-7920; (b) Allen, S.E.; Dokholyan, N.V.; Bowers, A.A. Dynamic Docking of Conformationally Constrained Macrocycles: Methods and Applications. ACS Chem. Biol. 2016, 11(1), 10-24.
- Watts, K.S.; Dalai, P.; Tebben, A.J.; Cheney, D.L.; Shelley, J.C. Macrocycle Conformational Sampling with MacroModel. J. Chem. Info. Model. 2014, 54(10), 2680–2696.
- Giordanetto, F.; Kihlberg, J. Macrocyclic drugs and clinical candidates: what can medicinal chemists learn from their properties? J. Med. Chem. 2014, 57(2), 278-295.
- MCauley, J.A.; Rudd, M.T. Hepatitis C virus NS3/4a protease inhibitors. Curr. Opin. Pharmacol. 2016, 30, 84–92; (b) Pillaiyar, T.; Namasivayam, V.; Manickam, M. Macrocyclic Hepatitis C Virus NS3/4A Protease Inhibitors: An Overview of Medicinal Chemistry. Curr. Med. Chem. 2016, 23(29), 3404-3447.
- (a) Johnson, T.W.; Richardson, P.F.; Bailey, S.; et al. Discovery of (10R)-7-amino-12-fluoro- 2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h] [2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem. 2014, 57(11), 4720-4744; (b) Basit, S.; Ashraf, Z.; Lee, K.; Latif, M. First macrocyclic 3rd-generation ALK inhibitor for treatment of ALK/ROS1 cancer: Clinical and designing strategy update of lorlatinib. Eur. J. Med. Chem. 2017, 134, 348-356.
- (a) William, A.D.; Lee, A.C.; Goh, K.C.; et al. Discovery of kinase spectrum selective macrocycle (16E)-14-methyl-20-oxa-5,7,14,26-tetraazatetracyclo[19.3.1.1(2,6).1(8,12)] heptacosa-1(25),2(26),3,5,8(27),9,11,16,21,23-decaene (SB1317/TG02), a potent inhibitor of cyclin dependent kinases (CDKs), Janus kinase 2 (JAK2), and fms-like tyrosine kinase-3 (FLT3) for the treatment of cancer. J. Med. Chem. 2012, 55(1), 169-196; (b) Verstovsek, S.; Komrokji, R.S. A comprehensive review of pacritinib in myelofibrosis. Future Oncol. 2015, 11(20), 2819-2830.
- (a) Cardote, T.A. Ciulli, A. Cyclic and Macrocyclic Peptides as Chemical Tools to Recognize Protein Surfaces and Probe Protein-Protein Interactions. ChemMedChem 2016, 11(8), 787-794; (b) Dougherty, P.G.; Qian, Z.; Pei, D.; Macrocycles as protein-protein interaction inhibitors. Biochem. J. 2017, 474(7), 1109-1125.
- Doak, B.C.; Zheng, J.; Dobritzsch, D.; Kihlberg, J. How beyond rule of 5 drugs and clinical candidates bind to their targets. J. Med. Chem. 2016, 59(6), 2312–2327.
- (a) Walensky, L.D.; Bird, G.H. Hydrocarbon-stapled peptides: principles, practice, and progress. J. Med. Chem. 2014, 57(15), 6275-6288; (b) Cromm, P.M.; Spiegel, J.; Grossmann, T.N. Hydrocarbon stapled peptides as modulators of biological function. ACS Chem. Biol. 2015, 10(6), 1362-1375.
- Chang, Y.S.; Graves, B.; Guerlavais, V.; et al. Stapled α-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110(36), E3445-E3454.
- Meric-Bernstam, F.; Saleh, M.N.; Infante, J.R.; et al. J. Clin. Oncol. 2017, 35(15 suppl.), 2505 (Oral presentation at 2017 ASCO Meeting, Chicago, IL, June 2017, Abstract 2505).
- Iyer VV. A Review of Stapled Peptides and Small Molecules to Inhibit Protein-Protein Interactions in Cancer. Curr. Med. Chem. 2016, 23(27), 3025-3043.