Introduction
Low Endotoxin Recovery (LER) has been THE hot industry topic for many years now and still there are debates on some fundamental points. Fortunately, the collective eff orts of regulatory authorities, biopharma manufacturers, industry groups, as well as instrument and reagent vendors have provided a much clearer direction over the last five years. This paper describes the Sanofi holistic approach to LER, which was generated by an internal group of risk management and endotoxin subject matter experts. The harmonized approach leverages the existing knowledge base and closes the gap in some key areas with a more comprehensive toolkit than previously described in the literature.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
This paper also intentionally avoids some of the ongoing debates regarding the “true mechanism” of LER, appropriate uses of Reference Standard Endotoxin/Control Standard Endotoxin RSE/CSE vs. Naturally Occurring Endotoxin (NOE), and potential medical or patient impact. Those topics are well documented in previous work and ongoing work from other authors and industry sub-teams.
The focus of this paper is a description of the practical toolkit to address LER for investigational medicinal products and previously registered commercial products. The design of the commercial product risk-based approach is described along with a summary of the use of the Sanofi Endotoxin Ingress Risk Assessment (EIRA). The study based investigational product approach is described along with an investigational product LER case study.
Risk-Based Approach for Registered Commercial Products
The principles of quality risk management as defined in ICH Q9, Quality Risk Management, were leveraged in the generation of the commercial product strategy. The approach was designed to dovetail with any existing microbial control strategies and supporting risk assessments. The Risk Assessment facilitates information-based decision mechanisms by evaluation of the current product and process-relevant risks, existing control effectiveness, and any harmacovigilance field signals. It was designed as a beneficial enhancement to mature proactive risk programs or as a standalone approach for developing risk programs.
Scope Determination
In order to determine which legacy commercial products are in-scope for deeper evaluation of LER four key elements are considered:
- Product type, route-of-administration, or potential endotoxin safety concern/testing
- Formulation components in the drug product
- Test method reagents/platform
- SME evaluation or other known factors that affect scope determination (e.g. possible non-classical LER mechanisms - masking)
Risk-Based Approach Overview
The Sanofi commercial products approach is a multi-step process that focuses on a customized risk assessment format developed specifically for LER called the Endotoxin Ingress Risk Assessment (EIRA). The EIRA was designed to assess the overall endotoxin ingress risk for in-scope product samples. Although the EIRA provides a mechanism to reach a final rating for the overall risk of endotoxin contamination, it should be supported by a technical report that documents supporting data, rationale, and justification for the ratings.
An example flow chart for the risk-based approach is provided below.
EIRA Overview
The EIRA is a structured evaluation of current potential risks, effectiveness of existing controls, and product field data. The EIRA team may include representatives from departments such as:
- Microbiology
- Manufacturing
- Risk Management
- Facilities
- Utilities
- Regulatory
- Quality Management
- Contamination Control
- Pharmacovigilance
The EIRA team rates the individual endotoxin ingress risk elements (numerical) and individual endotoxin ingress control elements (numerical) which are then compiled to arrive at a preliminary risk level (low, medium, high). Available product field data is then considered to arrive at an overall endotoxin ingress risk level (low, medium, high). The overall endotoxin ingress risk level is then compared to an action table for proposed mitigation activities.
A diagram of the EIRA design components is provided below:
The individual endotoxin ingress Risk Elements included in the assessment are:
- Hygiene Program
- Employee Training and Aseptic Technique
- Manufacturing Process – Open Processing and Room Classification
- Manufacturing Process – Clean (and/or sterilized)/Dirty Equipment
- Manufacturing Process Design
- Raw Materials and Components Testing
- Facility Design – Standing Water
- Facility Sanitization Disinfectant Efficacy
- Facility Design – Area Classification
- Facility Design - Maintenance and Facility Conditions
- Equipment and Components (Endotoxin Levels and Validation)
- Utilities – Water, Steam, Gas
- Utilities – Rouge and Passivation
The individual endotoxin ingress Control Elements included in the assessment are:
- Personnel – Hygiene, Training and Gowning
- Historical Product Stream Gram Negative Bioburden Results
- Raw Material and Components – Historical Performance
- Raw Material and Components – Supplier Performance
- Utilities Requalification Performance
- Environmental Monitoring Results
- Environmental Monitoring Performance
The Field Data included in the assessment is:
EIRA Execution Process, Details, and Examples
Step 1 - Rate the individual endotoxin ingress risk elements.
An example of the individual risk element rating for manufacturing process (open processing and room classification from the EIRA is provided below – lower scores represent lower risk):
Step 2 - Determine the Endotoxin Ingress Risk Level
Once all the other individual endotoxin risk elements (Hygiene Program, Employee Training and Aseptic Technique, etc.) are rated using their own corresponding scoring scale, the total is calculated and compared to a predefined scale, similar to the example in Table 1 below:
Step 3 - Rate Individual Endotoxin Ingress Control Elements
An example of the effectiveness of controls individual element rating for historical process stream Gram negative rod bioburden results is provided below. Lower scores correspond to higher ratings, representing more effective control performance:
Step 4 - Determine the Controls Effectiveness Level
Similar to the risk elements, after each of the other individual control effectiveness elements are rated using their own corresponding scoring scale. The sum of the ratings is calculated and compared to a pre-defined scale, similar to the example below in Table 2 below:
Step 5 - Determine the Preliminary Risk Level
The preliminary risk level is determined by using the Endotoxin Ingress Risk Level calculated from Table 1 and the Controls Effectiveness Level determined from Table 2. The intersection point of the levels on the grid in Table 3 below establishes the preliminary risk level.
Step 6 – Determine Individual Rating for Pharmacovigilance Review of Field Data
To determine if there are any signals from field, 12 months of data is reviewed by pharmacovigilance. Determine the level (Confirmed or Not Confirmed) for using Table 4.
Step 7 - Determine the EIRA Result
Using Table 5 determine the intersection of the Preliminary Risk Level from Table 3 and the individual rating for Pharmacovigilance Review of Field Data in Table 4. The intersection point is the EIRA Result (High, Medium, or Low). Record the EIRA Result in Table 5 below.
Step 8 - Determine the Recommended Risk Mitigation Actions
Using Table 6, identify recommended risk mitigation actions to reduce the risk to acceptable levels.
The justification for each individual rating within the EIRA described above must be supported and documented in an EIRA report. The ratings alone are not considered sufficient. Related existing microbial control risk assessments (e.g. HACCP) should be cross-referenced, as should any supporting microbial control strategies or applicable procedures.
The EIRA outcomes, risk mitigation, and justification provides an evaluation of the overall risk of endotoxin ingress for a given process and product. That information can then be used for leadership to make an informed decision and prioritize the appropriate next actions (e.g. risk mitigation, formal risk assessments, formal LER hold time studies, no formal LER hold time studies.)
Strategy for Investigational Medicinal Products
The strategy for investigational medicinal products is similar to the previously registered commercial product, however formal LER studies are required for all new BLA fi lings. Therefore an EIRA is not required for these products since there is no assessment required to instruct the appropriate path forward.
The approach for investigational medicinal products is summarized in the flow chart below:
LER Theoretical Likelihood Tool Overview
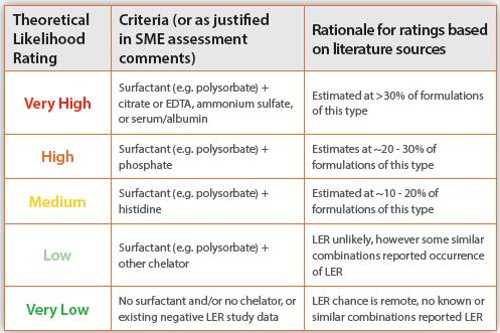
Although no universally accepted mechanism for LER has been identified, some theoretical estimations can be made. Components that are known potential contributors to “classical LER” are well documented in the literature. For example, a surfactant (e.g. polysorbate) combined with a divalent metal ion chelator (e.g. citrate, EDTA). Some “non-classical LER” masking factors have also been reported (e.g. serum, ammonium sulphate, uncommon pH levels). Subject matter expert (SME) evaluation of other product-specific factors or existing studies may also improve the estimation of the theoretical likelihood of LER. LER likelihood can help prioritize activities or direct more focus to certain products. The estimation of likelihood does not replace the requirement of formal LER studies for new products, regardless of the result of the evaluation. The basic theoretical LER likelihood rating tool design based on FDA presentations of drug sponsor BLA results, external experience, and internal Sanofi experience is provided in this chart:
LER Endotoxin Sample Hold Time Studies
LER related activities and studies must be planned and performed at the appropriate clinical stage to ensure successful outcomes. Feasibility studies are recommended during early stages to allow time for troubleshooting and remediation prior to formal studies and to ensure safety of future clinical batches.
In Phase 1, recommendations are to avoid known high theoretical LER likelihood formulations when possible and consider LER feasibility studies especially for high likelihood LER formulations. For many product types it is challenging to avoid divalent metal ion chelators and surfactants, but when possible the judicious selection of excipients and concentration can reduce the theoretical likelihood of LER.
In Phase 2, if not already completed, LER feasibility or formal studies should be performed depending on product strategy and timing. If known, the routine operations test conditions should be matched and study designs should follow best practices required for formal studies.
In Phase 3, formal LER studies matching routine operations test conditions are required. Supporting Rabbit Pyrogen Test and/or Monocyte Activation Test data must also be generated as required per regulatory requirements.
Principle
During LER/HT testing, undiluted samples are spiked with RSE or CSE. Samples are then held for a length of time in a container and at a temperature relevant to that particular test article and manufacturing process. At time points throughout the holding time, the sample is then diluted to the working dilution determined for routine testing and tested for endotoxin. The readout at time T is the percent CSE or RSE spike recovery in the sample, compared to the percent spike recovery in the LAL reagent water (LRW) negative control at the same hold time T. LER is diagnosed and confirmed if the CSE/RSE spike recovery in the sample is <50% for two consecutive time points. The demonstrated stability of assayable endotoxin content over time is the latest time point tested that the percentage of CSE/RSE spike recovered is strictly greater that 50%.
Case Study
The procedure for LER studies described above was applied to an investigational vaccine currently in Phase 3.
The DP (drug product) stage was selected for this assessment as the
Bacterial Endotoxin Testing (BET) is performed on this stage for release.
Theoretical Low Endotoxin Recovery Likelihood Tool
In accordance with the LER Risk Based Approach for investigational medicinal products described previously, the assessment using the Theoretical Low Endotoxin Recovery Likelihood Tool was performed for the vaccine DP in two steps:
1 - Evaluation of the formulation components using the theoretical LER likelihood assessment on the vaccine DP.
2 – Final theoretical likelihood rating is assigned based on the theoretical
LER likelihood tool.
Five potentially LER causing components were identified in the vaccine DP formulation:
- Surfactant
- Strong divalent metal ion chelator: EDTA
- Phosphate mono- and bivalent
- Weaker divalent metal ion chelator
- Serum/protein(s)
The final LER likelihood rating based on the evaluation using the Sanofi - developed likelihood tool for the vaccine DP was categorized as ‘Very High’ due to the presence of Surfactant and EDTA. FDA presented drug sponsor generated
BLA data indicates this combination of components aligns with an approximate 30% chance of LER when evaluated using CSE or RSE spike recovery studies.
Study Design
The LER study was conducted using the “Multi-aliquot” mode: one large sample volume and an identical LRW (Low Reagent Water) control were spiked with 5 IU/mL of RSE to target the mid-point of the standard curve.
They were then aliquoted at the start of the study (Day 0) on the same day prior to storage at +5°C±3°C in the vaccine product final containers.
Afterwards, analyses were performed on these aliquots at the selected time points as per our internal kinetic quantitative chromogenic LAL SOP and as per the conditions defined during the vaccine DP routine validation (diluent, standard range, working dilution, Lonza reagents).
Product was tested at Day 0, 1, 2, 7, 14, 22 and 30 after storage at +5°C±3°C.
Lot was tested as 3 replicates in duplicate (ie 6 times) by two analysts.
The table below summarizes the results of the study.
The percentage of average recovery of RSE did not drop less than 50% for two or more consecutive time points: the 3 replicates of the vaccine DP did not exhibit LER.
Assayable endotoxin content over time in undiluted product DP was demonstrated stable over 30 days at +5°C±3°C after sampling.
Discussion of Case Study Results
Per the likelihood tool, a very high likelihood (~30% chance) of LER was established for this vaccine DP. However, we demonstrated experimentally that this formulation does not exhibit LER. To better understand this phenomenon; the vaccine formulation can be further analyzed.
The phosphate content was comparably high in this vaccine formulation and there was a relatively low concentration of strong divalent metal ion chelator. On one hand, the presence of arginine may have blocked some phosphates and EDTA due to its positively charged amine groups. On the other hand, serum/protein(s) may have a stabilizing effect by absorbing surfactants or other amphiphilic compounds such as the type of surfactant used in this formulation. Thus, they could not form mixed, low-activity aggregates with LPS and endotoxin. Generally, certain proteins can promote LER by absorbing LPS or bivalent ions; others can inhibit LER by absorbing surfactants or chelating agents. It depends on the individual protein structure (charged and hydrophobic moieties on surface). Especially, the high concentration of proteins contained in our vaccine DP could be stabilizing against LER.
Taken together, the physico-chemical properties of the proteins were the suspects for the observed lack of LER.
It is important to remember that the estimation of LER likelihood described here is only one component of an overall holistic strategy designed to satisfy regulatory requirements. It does not replace the necessity of a formal LER study for new products, regardless of the result of the evaluation, as demonstrated here.
Conclusion
LER is currently a hot topic in the industry, but even highly theoretically likely LER formulations such as EDTA with a surfactant do not always result in experimentally confirmed LER. Properly designed and correctly interpreted experimental studies remain critical in determining if LER is present in a given formulation. Existing commercial products, with demonstrated low endotoxin ingress risk, documented effective controls, and strong patient safety records, can be appropriately managed using a holistic risk-based approach.
Holistic strategies, toolkits, and emerging guidance is finally advancing the LER topic past the theoretical debates and toward a common ground more useful for practical application.
Acknowledgements
Thank you to all (far too many to name) the risk and microbiological expert that participated in the Sanofi Quality Commission that was responsible for the generation of this harmonized holistic risk-based approach to LER.
We would like to warmly thank Frédéric VACHER, technologist in Analytical R&D Europe department, for his technical assistance in the case study presented here.
Finally, thank you to all of those who continue to contribute countless hours to LER industry sub-teams, task forces, research, literature articles, conference presentations, technical discussions, and even heated arguments. All of these are key building blocks for our collective progress on LER. Equal thanks to those referenced below and those who were not.
Authors
Mark Kapeckas’ career spans the fi elds of traditional pharmaceuticals, electronic materials, contract manufacturing, and biologics. Roles have included positions in R&D for test methods and new products, Project and Program Management, Quality Control, Quality Assurance, Quality Engineering. Mark has focused this unique experiential perspective on Biologics Quality for the past 10 years and currently leads several global initiatives within Sanofi , including the global LER program.
Marine Marius is Scientist in the Analytical Sciences department of Sanofi Pasteur. She leads the development, validation and implementation of rapid microbiology methods (bacteriology and molecular biology) for commercial and investigational products. She is an internal SME in endotoxin, mycoplasma and mycobacteria testing.
Jon Williams has worked in Quality Risk Management (QRM) for the Sanofi Biologics organization for the past 7 years where he supported manufacturing sites in the implementation and integration of QRM principles throughout the site quality organization including training on formal risk tools (FMEA, HACCP, etc.) and the development of custom risk tools. Jon’s current role is with the Sanofi Global Proactive QRM Program tasked with implementing a proactive QRM Program across the entire Sanofi organization by providing harmonized practices targeted at anticipating and avoiding unacceptable risks based on ICHQ9 (Risk Management) guidelines.
References
- Chen J.,- Genentech/ROCHE, “Low Endotoxin Recovery in Common Biologic Products”: Micro PDA Conference 2013
- Bolden J, Platco C, Dubczak J, Cooper J, McCullough KM. - 41(5) Stimuli to the Revision Process: “The Use of Endotoxin as an Analyte in Biopharmaceutical Product Hold-Time Studies”. Pharmacopeial Forum 2015
- Reyes Candau-Chacon, “Microbial control for biotech product drug substance manufacturing from an FDA perspective”, presentation date October 20th 2015
- Reich J., Reliability of Endotoxin-Detection Mechanistic Principles of Endotoxin-Masking and Strategies for De-Masking. Presented at the Parenteral Drug Association Conference, Berlin, Germany. February, 2014
- “Microbial Monitoring For Biological Drug Substance Manufacturing: An Industry Perspective”, PDA Journal and Pharmaceutical Science and Technology, Vol. 69, No. 3, May–Jun 2015: 450-460 (BPOG editorial)
- Stevens-Riley, “Ten Things that the FDA would like you to know about LER”, presentation date September 14th – 15th 2015
- Cooper, J., “LER: Microbiology’s Hottest Urban Myth”, American Pharmaceutical Review, November 30th 2015
- Hughes, P., “Endotoxin and Biotech Products: An FDA Perspective”, 10th Annual PDA Global Conference on Pharmaceutical Microbiology, October 20th 2015
- Hughes, P., “FDA Perspective on Endotoxin Testing and LER”, 11th Annual PDA Global Conference on Pharmaceutical Microbiology, Session A2, October 24th 2016
- Hughes, P., “FDA’s Current Thinking on Endotoxin Testing and LER”, PharmaLab, November 2017
- McCullough, K., “Current USP Perspectives on LER and Endotoxin Testing”, 11th Annual PDA Global Conference on Pharmaceutical Microbiology, October 2016
- Von Wintzingerode, F., “Low Endotoxin Recovery, A Brief Overview”, [on line]. Retrieved from https://www.americanpharmaceuticalreview.com/Featured-Articles/239898-Low-Endotoxin-Recovery-A-Brief-Overview/, September 30, 2016