Abstract
Dissolution technique is an excellent tool for monitoring batch-to-batch quality control and to understand drug release in in-vivo conditions. Dissolution methods are mostly recommended for solid oral dosage forms. However, they have also been extended to other dosage forms such as suspensions, transdermal patches, chewing gums, etc. Choosing the right in-vitro method and apparatus provides an excellent view of the formulated dosage form. Over the past two decades, efforts towards harmonization between pharmacopoeias have led to more uniform methods for in-vitro dissolution of conventional dosage forms across the board. Dissolution studies for novel drug delivery systems have been limited primarily to literature citations and are yet to find a place in the pharmacopoeia.
Introduction
Dissolution is defined as the process by which a solid substance enters into a solvent to yield a solution and is controlled by the affinity between the solid substance and the solvent. Dissolution testing is an official test in the pharmacopoeia for evaluation of a variety of dosage forms (solid, semi-solid, suspension and transdermal). However, no standard method is recommended for in-vitro release testing of novel dosage forms such as microspheres, nanoparticles, liposomes, aerosols and drug eluting stents (DES). Developing a standardized dissolution method for these dosage forms is difficult because the release is dependent on various physico-chemical properties of the formulations, and the biological environment in which the drug release will occur. The objective of this article is to familiarize the readers with dissolution methods available or followed for testing of some of these novel dosage forms.
Dissolution testing is harmonized between United States Pharmacopoeia (USP), European Pharmacopoeia (EP) and Japanese Pharmacopoeia (JP). The general descriptions of USP apparatus 1 to 4 and their suitability, acceptance criteria of the tests are given in USP and EP. USP apparatus 1 and 2 are described as is in the JP. While USP apparatus 3 is not accepted in JP, USP apparatus 4 is described as apparatus 3 in the JP. A mastication instrument for evalation of chewing gums is also detailed in EP. Dissolution test for transdermal systems is described in USP and EP, while there is no dissolution test specified for transdermal patches in JP.
Microspheres
Microspheres are submicron particulate drug delivery systems which have no in-vitro dissolution technique described in any pharmacopoeia. The dissolution techniques used either for research work or commercial purpose depends on how much they are able to discriminate the drug release profile using the technique. The United States Food and Drug Administration (USFDA) looks specifically for a method which can discriminate the drug release and provide an in-vitro/in-vivo correlation (IVIVC). The methods described by the innovator are mostly classified and very little information is available in the public domain. Though the number of microsphere-based products has grown, no specific guideline has been framed for the dissolution studies. In a recent article, Kumar and Palmieri have provided a drug product specification for polymeric microspheres [1].
Most of the microsphere products are administered by parenteral route as a suspension either through the intramuscular or subcutaneous route. Based on the site and route of injection, the drug release profile of the injected microspheres may differ. This is because of the difference in pH at the site of injection, volume of fluid available and the mechanism in which the drug is being released from the microspheres. Primarily, drug release from polymeric microspheres is by erosion-based diffusion. Polymeric erosion depends on hydrolysis of the polymer. Polymers with different molecular weight show different pattern of erosion and also depends on the site of injection. This can be explained with the example of Lupron Depot® (for once a month injection) which is prepared using a polymer that needs to release all its drug content within a one month period while Lupron Depot® (for once in three months injection) is composed of a different polymer so as to release the drug in three months.
Table 1 - Typical Example of Release Specifications for Once a Month Microsphere Formulation

Typically, the in-vitro drug release profile is carried out for the drug product to see if the drug is released over the desired period of time. However, in the case of microspheres, the drug release ranges from weeks to months. Performing in-vitro drug release profile for the specified time period (e.g., 3 months) and to then release the batch for commercial use is time consuming and unrealistic. In order to overcome such problems, in-vitro release is carried out under accelerated conditions where, only the drug release is accelerated without affecting the mechanism by which the drug is released. In general, the specification is set based upon the in-vivo responses (effects and side-effects). Initial or burst release of the drug at the site of injection immediately following administration may be necessary to elicit the initial pharmacological response. However, too much drug release at this stage may have adverse effects. Table 1 describes a model of what the specification can look like for once a month microsphere product.
Table 2 - In-vitro Dissolution of Indomethacin Microspheres using Different Dissolution Techniques
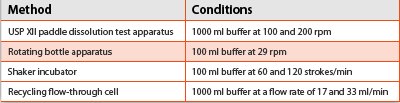
In-vitro release of microspheres has been studied using different techniques. Table 2 describes an example for in-vitro dissolution of indomethacin-containing microspheres [2].
In-vitro dissolution using dialysis techniques, involve the use of a dialysis membrane bag of certain molecular weight cut off (MWCO). The microsphere suspension is placed in the dialysis membrane bag, sealed from both ends and suspended in the buffer under constant agitation using a shaker or paddle [3]. The volume of the microparticulate suspension is 5–10 times less than the bulk media in order to maintain sink conditions. It offers several advantages like ease of sample withdrawal, physical separation of microparticles from buffer, and convenience. However, this technique cannot be used if the drug binds to the dialysis membrane.
Nastruzzi et al. [4] studied the release of bromocriptine mesylate from microspheres using dialysis tubes and a flow-through cell method and compared the reproducibility between the two methods. Dialysis technique exhibited more drug release with longer time to plateau whereas with the flow-through cell, the time to reach the plateau was comparatively shorter, and lesser amount of drug was released.
A modified flow through cell technique is also reported where the microspheres were mixed with glass beads in the cells [5]. This modification aided in preventing the aggregation of microparticles, decreased dead volume and increased laminar flow in the flow through cells. All the investigated methods are associated with merits and demerits. Among the various techniques available, dialysis method seems to be more useful. Some of these techniques also hold good for studying the in-vitro release of nanoparticles.
Nanoparticles
Assessment of drug release from nanoparticulate systems is critical because the efficiency of the delivery system is dependent on this important quality control parameter. Various methods available to assess the drug release from nanoparticulate systems can be broadly divided into 3 categories (i) membrane diffusion such as dialysis, reverse dialysis and diffusion cell; (ii) sample and separate using centrifugation, filtration and centrifugal ultrafiltration technique; (iii) in-situ measurement e.g., UV spectrometry, fluorescence spectrometry and differential pulse polarography.
In the in-vitro dissolution using the bottle method, nanoparticles are stirred in bottle containing dissolution media and maintained in water bath at 37ºC for a definite time. Samples are withdrawn and separated by ultra centrifugation for subsequent analysis. In other methods which are similar to this, the sample withdrawn can be treated by ultrafiltration or diffusion cells separated by membranes.
Each of these methods suffer from certain drawbacks; like passage of drugs across the membranes can be a rate-limiting step in membrane methods; while the high-speed ultracentrifugation in sample and separate method can affect the drug release. These factors might result in false estimation of the drug release. Hence, the selection of the appropriate method is important. The advantage of in-situ methods is that the nanoparticles are incubated in release media and analyzed in the media without separation or avoiding loss of drug during sampling.
Hellea et al [6] designed a novel on-line system for evaluating the drug release from indomethacin and beclomethasone dipropionate nanoparticles. In this method, nanoparticles are packed into small empty vessel which is connected to multiport modulation valve attached to an HPLC system. The modulation valve has two loops. The eluent from the vessel, packed with nanoparticles is continuously collected and transferred to the analytical LC column for separation of analytes. This setup allowed faster monitoring of drug release.
Liposomes
Parenteral route enables rapid onset of action but suffers from patient discomfort. To overcome such difficulties, prolonged release injectable liposomal formulations Doxil®, DepoCyt®, AmBisome® and DaunoXome® have been developed. These products release for extended periods of time ranging from several hours to days or weeks. Most of them are administered by intravenous or subcutaneous route. The methods that have been reported for in-vitro dissolution of liposomes include sample and separate, flow through cell, dialysis, reverse dialysis, micro dialysis and using Franz diffusion cells. Most of these methods are not standardized, and hence, the results obtained are highly variable.
In reverse dialysis technique, dialysis membranes of known MWCO were filled with 2 ml buffer. These membranes were suspended in glass tubes filled with bulk volume of buffer. Liposomal suspension (2 ml) was added to the media outside the dialysis bag. At specified time intervals, the dialysis bag was removed and samples were analyzed [7].
Bharadwaj and Burgess [7] developed and utilized a novel dialysis adapter that can be used with USP dissolution apparatus-4. This modified apparatus was used to study the release of dexamethasone liposomes from different formulations. The technique may be applied for in-vitro release testing of colloidal disperse systems such as nanosuspensions, liposomes and emulsions.
A few unique membrane-free methods for in-vitro testing of liposomes have also been demonstrated [8]. The release profile can be computed irrespective of the viscosity and probable phase transitions of the sample. These techniques were named as single drop and inverted cup technique. In an inverted cup technique, a hole is drilled in the bottom of the cup made of borosilicate glass tube. Sample is introduced into the cup through this opening and air is vented out. The sample is suspended by buoyancy which either spreads out or sticks to the glass based on the polarity of the sample.
Summary
This article summarizes in brief the techniques followed by different researchers to evaluate the in-vitro release testing of novel dosage forms. These methods provide valuable insights for development of standardized testing methods for evaluation. The shortfalls in most of these methods can be overcome by scientific interactions with researchers working in this field around the globe. These discussions form the basis for revising the guidelines for evaluating novel dosage forms.
References
- R. Kumar and M.J. Palmieri Jr. AAPS J. 12(1) (2009) 27-32.
- B. Conti, I. Genta, P. Giunchedi, and T. Modena, Drug Dev. Ind. Pharm. 21(1995).1223-1233.
- S. S. D’Souza and P.P. DeLuca, Pharm. Res. 23 (2006) 460 – 474.
- C. Nastruzzi, E. Esposito, R. Cortesi, R. Gambari et al., J. Microencapsul. 11 (1993) 565 – 574.
- B.S. Zolnik, J.L. Raton, and D.J. Burgess, Diss. Technol. 12 (2005) 11-14.
- A. Hellea, S. Hirsjärvi, L. Peltonen, and J. Hirvonen, J. Pharm. Biomed. Anal. 51 (2010) 125–130.
- U. Bhardwaj and D. J. Burgess, Int. J. Pharm. 388 (2010) 287–294.
- L. Söderberg , H. Dyhre, B. Roth, and S. Björkman, J. Control. Rel. 113 (2006) 80–88.
Author Biographies
Guru V. Betageri, Ph.D. is Professor of Pharmaceutical Sciences and Associate Dean of Graduate College of Biomedical Sciences at Western University of Health Sciences. Dr. Betageri’s expertise is in the area of formulation and development of drug delivery systems with emphasis on lipid-based delivery systems. Dr. Betageri has published more than 60 original research papers in reference journals and presented more than 100 papers at National and International meetings.
Veeran Gowda Kadajji, Ph.D. is a Post-Doctoral Researcher currently working at Western University of Health Sciences, Pomona, CA. He has 3 years of experience in the Pharmaceutical industry. His research interests are formulation & characterization of novel oral drug delivery systems. He has authored several manuscripts and serves as a reviewer for many journals.
Natarajan Venkatesan, Ph.D. is an Assistant Professor at Chicago College of Pharmacy, Midwestern University, IL. Until recently, he was a post-doctoral fellow at Western University of Health Sciences, Pomona, CA. He has about 8 years of post-doctoral experience in Japan and US in addition to 3 years of Industrial R&D experience. He has published in peer-reviewed journals and is co-inventor in a handfull of patent applications.
This article was printed in the September/October 2011 issue of American Pharmaceutical Review - Volume 14, Issue 6. Copyright rests with the publisher. For more information about American Pharmaceutical Review and to read similar articles, visit www.americanpharmaceuticalreview.com and subscribe for free.