Abstract
Solid form screening is commonly performed to find a candidate with optimal properties for early development or to find a form with different properties to improve a formulation in later development. A variety of screens can be performed including polymorph, salt, co-crystal, amorphous, and amorphous dispersion. X-ray powder diffraction (XRPD) is commonly used at various stages of screening to identify and characterize new forms. It is also used to help evaluate other properties, such as physical stability and manufacturability, in order to choose the best form for development. This paper discusses the use of XRPD during screening and form selection of pharmaceutical materials.
Introduction
It has been reported that up to 89% of pharmaceutical compounds screened result in more than one solid form [1]. Studies have shown that these different solid forms can exhibit diverse properties, such as solubility [2], bioavailability [3], and manufacturability [4], which will influence the targeted drug product and significantly impact the development of a compound. Understanding the possible solid forms available can help direct successful processing of both the drug substance and drug product resulting in robust and consistent manufacturing. Screening is a recognized method for finding new forms of both crystalline and amorphous materials and numerous studies have been reported on various methodologies and characterization techniques employed in these studies [5]. X-ray powder diffraction is a common technique to determine if a new form has been found since it gives direct information on the packing of the molecule in the solid state and can be referenced back to the single crystal structure of the form. This article will discuss the role of solid form screening and selection during drug development and how XRPD is used in these pivotal studies.
Forms
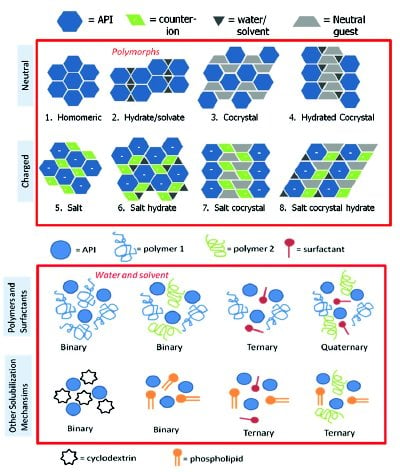
Figure 1 - Classes of multicomponent pharmaceutical materials. (a) Schematic of crystalline materials showing neutral and charged species. The red box indicates polymorphs are possible for all the multicomponent crystals contained within the box (adapted from Reference 7). (b) Schematic of amorphous solid dispersions showing binary, ternary, and quaternary possibilities for polymers and surfactants. Other solubilization techniques using cyclodextrins and phospholipids are included for completeness but have a different mechanism for solubilization when compared to polymer and surfactant systems. The red box indicates that properties can change with water or solvent content.
There are a number of solid forms available for pharmaceutical development and these can include crystalline forms (polymorphs, salts, co-crystals) and amorphous materials (amorphous drugs, amorphous solid dispersions). For crystalline materials, the solid forms of a compound can include multiple anhydrates, hydrates, and solvates, shown in Figure 1. For salts and co-crystals, it can include entities with different counterions/guests to form binary systems or the addition of water/solvent to form a ternary system [7]. Some co-crystals can be in a ternary system containing a salt and neutral component, commonly called salt co-crystals [7]. Add water or solvent to the salt co-crystal, and a quaternary system is now available. Different packings (polymorphs) are possible for all these forms resulting in a wide variety of crystalline materials available for development. For amorphous solid dispersions, a polymer is usually added to an amorphous API to form a binary system, but ternary and quaternary systems with multiple polymers and surfactants have also been reported [8]. Systems using different solubilization mechanisms, such as cycodextrins and phospholipids [9], are sometimes called amorphous dispersions as well. When the amorphous and crystalline possibilities are combined, it results in a wide landscape for screening and selection.

Figure 2 - Example of amorphous and crystalline forms of AMG 517: (a) 15:85 AMG 517:HPMCAS amorphous solid dispersion; (b) AMG 517 amorphous free base; (c) AMG 517 cinnamamide co-crystal; (d) AMG 517 free base form B; (e) AMG 517 free base form C (adapted from reference 11).
X-ray powder diffraction is one of the most common techniques for verifying different solid forms of a material [10]. It can differentiate amorphous materials from crystalline and in most cases readily exhibits differences between different crystalline forms based on the peak positions and intensities observed in the powder pattern, as shown in Figure 2 for different crystalline and amorphous forms of AMG 517 [11]. XRPD readily distinguishes between different forms of the free base (Forms B and C) as well as the AMG 517:cinnanamide co-crystal. Differences between the AMG-517 amorphous free base and the 15:85 AMG 517: HPMCAS amorphous solid dispersion are also readily apparent based on the shapes of the amorphous halos. This example illustrates the ease of using XRPD to initially identify different forms. Once it is determined that a different form is present, other methods, such as thermal and spectroscopic techniques (IR, Raman, solid-state NMR), can be used to gain additional information on the new form including solvation state, melting point, thermal transitions, and bonding interactions.
Screening
Screening for polymorphs [1,5] has been performed for many years and is a common practice in most companies. Salt screening [6] has routinely been used to find possible salts of a compound especially when trying to change properties such as solubility or stability. Co-crystal [6] and amorphous solid dispersion screening [3,8] are relatively new to the industry, but have the same basic steps as the other screens.

Figure 3 - Schematic of the screening and selection processes.
It is important to differentiate between form screening and selection as shown in Figure 3. Screening involves using various conditions to cover a wide landscape of possibilities with the goal of finding new forms. XRPD data are commonly collected for any solid samples that are generated, and these patterns are used to determine if new forms have been produced. Since a large number of samples are generated during a screen, available characterization data are limited for any new forms during the screen. This process provides information on the forms that are possible, but makes selecting a form difficult due to a lack of essential development information on the solid, such as solubility, stability, or hygroscopicity. To select a form it is important to produce more material, collect additional data on properties relevant to the project, and choose the best solid for your development needs.
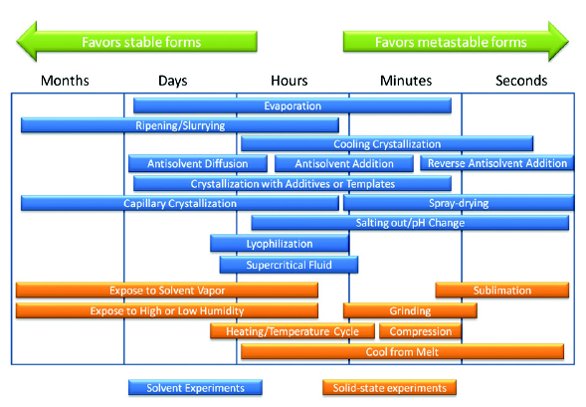
Figure 4 - Types of screening experiments (adapted from reference 12).
A number of different crystallization methods can be used in a screen, as shown in Figure 4. Solvent methods are the most common, but techniques stressing the solid under different conditions, such as drying or grinding, should also be included in many screens. By varying the conditions, a wider crystallization space is sampled which will help find new forms. It should be emphasized that a screen is a search for seeds and many methods may not be directly scalable into a process. When a new form is found on small scale, the seeds can many times be used to produce the form at a larger scale from a more conventional method. Many solid state screening techniques (heating, grinding, etc.) also mimic common processes that will be encountered during API or drug product manufacturing, enabling the screen to help predict problems early in development and guide manufacturing process selection.
It is important to realize that some crystallization techniques will favor metastable forms while others will tend to give thermodynamically stable forms. Fast crystallization methods will commonly result in metastable forms by trapping the unstable state during the crystallization process. These techniques are represented on the right side of Figure 4 and can be used to produce metastable or amorphous materials. Slow crystallization methods will commonly result in stable forms as represented on the left side of Figure 4. Solubility of the starting material also needs to be taken into account for screening studies. For solvents where the drug is highly soluble, a saturated solution may gel or result in an oil, therefore a fast or slow precipitation may not be the best choice, but an antisolvent addition may be a possibility. For solvents where the drug is very poorly soluble, heating the solution to increase solubility and cooling the solution at different rates may be a viable experiment. Slurry experiments can be very useful at concentrations around or greater than 3 mg/mL and a stable form screen has been suggested using this method [13]. Starting with estimated solubility measurements can help tailor the crystallization experiments to produce more solid samples during the screen.
Screening can be performed by manual, automated, or a combination of the two methods [1,14,15]. Manual screening involves samples set up in individual vials or containers under a variety of conditions including both solvent and solid-state methods. Automated screening involves a plate-like arrangement which can include small vials or wells. Plate screens usually involve only solvent-based methods such as solvent removal or anti-solvent addition. A large number of solvents can be explored using automated methods, but depending on the solubility of the drug in the solvents, a poorly soluble drug may not produce a high number of solid samples or will result in a sample size that is too small for analysis. A combination of automated and manual methods is suggested for most screens to cover the largest crystallization space.
Salt, co-crystal, and amorphous solid dispersions screens have a second component (counterion, guest, and polymer, respectively) to consider when determining the experimental conditions. Solubility of the API and the second component in the same solvent or miscible solvents must be considered. Non-solvent methods, such as melt techniques, are also used for these types of systems.
XRPD is commonly used as the initial method of analysis for form screens. For polymorph, salt, and co-crystal screens XRPD is used to determine if a new form has been produced by comparing the powder pattern to all known forms of the API and the counterion/guest. If a new form is found by XRPD, additional characterization by other methods is in order. For amorphous solid dispersion screens, XRPD is used to confirm a lack of crystallinity indicated by an amorphous halo in the powder pattern. The halos will move depending on the concentration and interactions of the API and polymer. Computational methods have also been used with XRPD data to establish miscibility of amorphous solid dispersions [16].
When analyzing samples from a manual screen, routine sample holders that accommodate the appropriate sample size are used for analysis. Automated screens with arrays of samples, such as plates, may require a special diffractometer configuration to accommodate sample analysis in the confined spaces of the multiple samples and to acquire representative data on a small sample size, which can be as small as 1-2 mg. Issues such as poor signal-to-noise, strong backgrounds, and inherent preferred orientation effects need to be addressed for screening samples, especially in plate configurations [15]. Manual and automated screens produce large numbers of powder patterns which need to be analyzed, with automated screens sometimes generating thousands of samples. Data analysis methods are needed to compare patterns and identify possible new forms. Due to the large number of patterns generated, these analysis methods are often automated. For example, techniques to group similar patterns are available such as cluster analysis, dendograms, and metric multi-dimensional plots [15]. Visual aids, such as color coding wells in a schematic of the plate based on the form produced, are also used to readily group forms and conditions. Once possible new patterns are identified, it is essential to compare them against known and new patterns to determine if mixtures are present. Many times the patterns can be indexed to identify a pure form. Preferred orientation effects can be assessed by comparing the experimental powder pattern to a calculated powder pattern from a single crystal structure if it is available. Comparing solution NMR data on possible new forms to the original material is helpful to ascertain if the molecule is intact or has undergone degradation during the screening process. Additional characterization of new forms including solvent content using thermogravimetry (TG) or gas chromatography (GC), melting point using differential scanning calorimetry (DSC) or hot-stage microscopy, and possible interactions using spectroscopic methods can also help determine if a new powder pattern represents a new form or a mixture of forms.
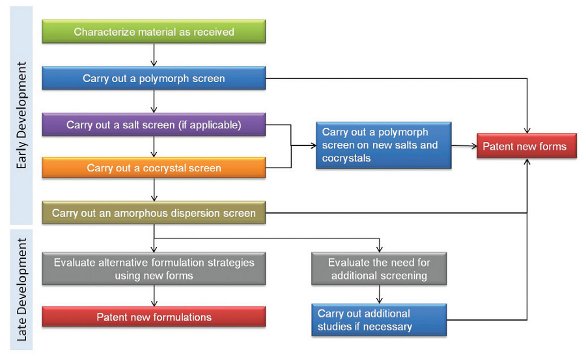
Figure 5 - Screening strategies throughout the drug development process.
Screening strategies will depend on the stage of development and more than one screen may be necessary throughout the life of the drug as illustrated in Figure 5. In early development, either the most stable or most soluble form may be desired for early animal and clinical studies. As development continues, additional screening may be needed to investigate forms for improved drug products. These studies may include a variety of screens to find the form with the desired properties. It is important to note that if a salt or co-crystal is chosen for development, a polymorph screen of the selected salt or co-crystal will also be needed to determine the most stable form as well as other forms that may be produced during processing.
Form Selection
A variety of factors need to be considered when selecting a form and these factors will be different for each molecule. The development plan needs to be taken into account, such as the type of dosage form, drug loading, processing conditions, etc. In addition, the properties of the forms need to be evaluated in order to select the best form for development. These can include solubility, physical and chemical stability, hygroscopicity, handling, flow, and manufacturability. These are in addition to traditional parameters for the drug substance such as purity, yield, impurity levels, morphology, and particle size.
X-ray powder diffraction plays a key role in property determination of new forms. Form changes need to be investigated and understood early in development in order to develop robust crystallization and formulation processes that will consistently produce the desired form [17]. Many times there is a disconnect between the screening experiments and the application to real life processes. Screening experiments can provide key information to help guide property determination and processing if it is reviewed and used effectively. Properties that will be considered here for illustration are physical stability, solubility, and formulation process development.
Stability studies are commonly performed for new drug entities with chemical stability and impurity formation being investigated. It is also important to monitor the physical stability under these same conditions to anticipate any form changes that may occur. As an example, many hydrates will dehydrate to a lower hydrate or anhydrous form at elevated temperatures. Anhydrous materials can also undergo form transformations to other anhydrous forms upon heating. These types of changes can be monitored using heating studies in an oven with subsequent XRPD analysis or in-situ variable temperature XRPD can be used to look for changes. In other cases, anhydrates will convert to hydrates or the API in an amorphous solid dispersion may crystallize under elevated relative humidity (RH) conditions. Equilibration in RH chambers with subsequent analysis by XRPD or in-situ variable RH XRPD experiments can be used to readily identify these form changes. Once the effect of temperature and RH on form changes is understood, this can be factored into other processes such as drying, formulation, storage, and packaging [17].
Solubility is an important parameter for new molecules especially with the emergence of many poorly soluble compounds in the drug discovery and development pipeline. Polymorphic forms can exhibit solubility differences that vary within a factor of 1-5, amorphous solid dispersions show an improvement one or two orders of magnitude higher, and salts and co-crystals fall between these extremes [18]. A comparison of solubility values of pure forms will provide important information when deciding on a solid form or dosage form. X-ray powder diffraction will allow identification of pure forms for these types of measurements. However, form changes during solubility and dissolution experiments are also possible and need to be investigated. Solids remaining at the end of solubility and dissolution experiments should always be analyzed initially by XRPD to determine if a form transformation has occurred under these conditions. If a form change has occurred, XRPD patterns can be compared to known forms (polymorphs, hydrates, salts, free acid/base) in order to identify the solids remaining. If a pattern is obtained that does not correspond to known forms, complementary methods will be needed to determine properties such as hydration state or a change in stoichiometry as would be observed from breaking a salt and forming the free acid/base or the formation of salts in buffered solutions.
Important formulation steps may need to be considered when selecting a form, such as suspension in a liquid, milling, wet granulation, or compression. Many times screening experiments can provide information on whether these steps will be an issue and cause process induced transformations [17]. If there is a possibility of form transformation, additional studies should be performed to help determine boundary conditions that reproducibly produce the desired form. XRPD data should be collected before and after small-scale experiments to identify form changes. Similar data can be collected during scale-up and correlated to in-process techniques such as NIR or Raman spectroscopy to ensure robust procedures as described in the Quality-by-Design (QbD) paradigm [19].
Once data are available, they need to be analyzed in order to choose the best form for development. A number of tools are available to assess the properties and find the best form for development. A common method is a flow chart or decision tree that will help narrow the choices based on the properties desired for development [20]. While general flow charts are available from a variety of sources, it is helpful to tailor the decision tree to the goals of the specific project. Many factors can be incorporated into the decision making process such as crystallinity, deliquescence/hygroscopicity, solubility, and physical stability for an oral dosage form. Other requirements may be needed for an alternate dosage form. An injectable formulation may need other properties such as pH solubility and stability or production of an acceptable lyophilized cake and these properties can be incorporated into an appropriate flow chart. A form matrix is another tool that can be used to evaluate different forms [21]. Materials and properties are grouped into a table and acceptable properties can be highlighted to find the solid form with the best properties.
It should be noted that a form with all the desired characteristics may not be available for every compound. When this is the case properties need to be evaluated based on the needs of the project and other solutions need to be assessed [22]. If the solid is sensitive to moisture or solvent, a dry or melt granulation should be considered as well as special packaging. If an undesirable transition occurs during compression, a capsule instead of a tablet may be a feasible option. Alternative approaches during processing, formulation, and packaging will commonly need to be explored and can be used to counter the less than ideal properties of the solid.
Conclusions
X-ray powder diffraction is a front line technique in solid form screening and selection based on its ability to give a fingerprint of the solid-state structure of a pharmaceutical material. Understanding the solid forms of a pharmaceutical compound provides a road map to help direct a variety of development activities ranging from crystallization, formulation, packaging, storage, and performance. Different screening and selection strategies are warranted in early and late development because different information is needed at the various stages. Solid form selection and formulation approaches need to be investigated together and tailored to the situation. It is important to include solid form selection and possible changes in form as part of the risk management strategy throughout the drug development process.
References
- G. P. Stahly, “Diversity in Single- and Multiple-Component Crystals. The Search for and Prevalence of Polymorphs and Co-crystals”, Crystal Growth and Design, 2007, 7, 1007-1026.
- S.Bassavoju, S. Bostrom, S. P. Velaga, “Pharmaceutical Co-crystal and Salts of Norfloxacin”, Crystal Growth and Design, 2006, 6, 2699-2708.
- D. Engers, J. Teng, J. Jimenex-Novoa, P. Gent, S. Hossack, C. Campbell, J. Thomson, I. Ivanisevic, A. Templeton, S. Byrn, A. Newman, “A Solid-State Approach to Enable Early Development Compounds: Selection and Animal Bioavailability Studies of an Itraconazole Amorphous Solid Dispersion”, Journal of Pharmaceutical Sciences, 2010, 99, 3901-3922.
- L. M. Katrincic, Y. T. Sun, R. A. Carlton, A. N. Diederich, R. L. Mueller, F. G. Vogt, “Characterization, Selection, and Development of an Orally Dosed Drug Polymorph from an Enantiotropically Related System”, International Journal of Pharmaceutics, 2009, 366, 1-13.
- Polymorphism in the Pharmaceutical Industry, Edited by R. Hilfiker, Wiley-VCH, Weinheim, 2006.
- A. W. Newman, S. L. Childs, B. A. Cowans, “Salt and Co-crystal Form Selection” in Preclinical Development Handbook, John Wiley and Sons, Hoboken, 2008, 455-481.
- S.L. Childs, L.J. Chyall, J. T. Dunlap, V. N. Smolenskaya, B.C. Stahly, G.P. Stahly, “Crystal Engineering Approach to Forming Co-crystals of Amine Hydrochlorides with Organic Acids. Molecular Complexes of Fluoxetine Hydrochloride with Benzoic, Succinic, and Fumaric Acids”, Journal of the American Chemical Society, 2004, 126, 13335-13342.
- B.E. Padden, J.M. Miller, T. Robbins, P.D. Zocharski, L. Prasad, J.K. Spence, J. LaFountaine, “Amorphous Solid Dispersions as Enabling Formulations for Discovery and Early Development”, American Pharmaceutical Review, 2011, Jan/Feb, 66-73.
- (a) M.S. Nagarsenker, R.N. Meshram, G. Ramprakash, “Solid Dispersion of Hydroxyopropyl-cyclodextrin and Ketorolac: Enhancement of In-vitro Dissolution Rates, Improvement in Anti-inflammatory Activity and Reduction in Ulcerogenicity in Rats”, Journal of Pharmacy and Pharmacology, 2000, 52, 949-956. (b) M. Fuji, K. Harada, M. Matsumoto, “Physicochemical Properties of Phenobarbital Solid Dispersions with Phosphatidylcholine”, Chemistry and Pharmaceutical Bulletin, 1990, 38, 2237-2241.
- XRPD pharma ref. H.B. Brittain, “X-Ray Powder Diffraction of Pharmaceutical Materials”, American Pharmaceutical Review, 2002, 5, 74-80.
- (a) M.K. Stanton, R.C. Kelly, A. Colletti, Y.-H. Kiang, M. Langley, E.J. Munson, M.L. Peterson, J. Roberts, M. Wells, “Improved Pharmacokinetics of AMG 517 Through Co-Crystallization Part 1: Comparison of Two Acids with Corresponding Amide Co-Crystals”, Journal of Pharmaceutical Sciences, 2010, 99, 3769-3778. (b) M. Kennedy, . Hu, P. Gao, L. Li, A. Ali-Reynolds, B. Chal, V. Gupta, C. Ma, N. Mahajan, A. Akrami, S. Surapaneni, “Enhanced Bioavailability of a Poorly Soluble VR1 Antagonist Using an Amorphous Solid Dispersion Approach: A Case Study”, Molecular Pharmaceutics, 2008, 5, 981-993.
- C. Anderton, “A Valuable Technique for Polymorph Screening”, American Pharmaceutical Review 2007, March/April, 34-40.
- J.M. Miller, B.M. Collman, L.R. Greene, D.J.W. Grant, A.C. Blackburn, “Identifying the Stable Polymorph Early in the Drug Discovery-Development Process”, Pharmaceutical Development and Technology, 2005, 10, 291-297.
- J. Aaltonen, M. Alleso, S. Mirza, V. Koradia, K.C. Gordon, J. Rantanen, “Solid Form Screening – A Review”, European Journal of Pharmaceutics and Biopharmaceutics, 2009, 23-37.
- R. Storey, R. Docherty, P. Higginson, C. Dallman, C. Gilmore, G. Barr, W. Dong, “Automation of Solid Form Screening Procedures in the Pharmaceutical Industry – How to Avoid the Bottlenecks”, Crystallography Reviews, 2004, 10, 45-56.
- A. Newman, D. Engers, S. Bates, I. Ivanisevic, R. C. Kelly, G. Zografi, “Characterization of API : Polymer Mixtures Using X-ray Powder Diffraction”, Journal of Pharmaceutical Science, 2008, 97(11), 4840-4856.
- K.R. Morris, U.J. Griesser, C.J. Eckhardt, J.G. Stowell, “Theoretical Approaches to Physical Transformations of Active Pharmaceutical Ingredients during Manufacturing Processes”, Advanced Drug Delivery Reviews, 2001, 48, 91-114.
- (a) M. Pudipeddi, A.T.M. Serajuddin, “Trends in Solubility of Polymorphs”, Journal of Pharmaceutical Sciences, 2005, 94, 929-939. (b) Hancock and Parks, “What is the True Solubility Advantage for Amorphous Pharmaceuticals?”, Pharmaceutical Research, 2000, 17, 397-404.
- L.X. Yu, “Pharmaceutical Quality by Design: Product and Process Development, Understanding, and Control”, Pharmaceutical Research, 2008, 25, 781-791.
- S. R. Byrn, R. R. Pfeiffer, and J. G. Stowell, Solid-State Chemistry of Drugs, 2nd edition, SSCI, Inc, West Lafayette, IN 1999.
- S.X. Yin and J.A. Grosso, “Selecting and Controlling API Crystal Form for Pharmaceutical Development-Strategies and Processes”, Current Opinion in Drug Discovery and Development, 2008, 11, 771-777.
- G. Z. Zhang, D. Law, E.A. Schmitt, Y. Qiu, “Phase Transformation Considerations during Process Development And Manufacture of Solid Oral Dosage Forms”, Advanced Drug Delivery Review, 2004, 56, 371-390.
Author Biography
Dr. Ann Newman is a pharmaceutical consultant at Seventh Street Development Group. She received her Ph.D. in Chemistry from the University of Connecticut. Dr. Newman started her career at Bristol-Myers Squibb where she performed characterization studies on drug substances and products. As Vice President of Materials Science at SSCI, Inc. she was involved in characterization, form screening/selection, quantitation, and problem solving for the pharmaceutical industry. As Vice President of Research and Development at Aptuit, she instituted a company-wide R&D initiative covering API, preclinical, formulation, solids, analytical, clinical packaging and regulatory. She holds an adjunct faculty position in Industrial and Physical Pharmacy at Purdue University and is author/collaborator on over 40 publications, 75 technical presentations, and 40 webinars.
This article was printed in the September/October 2011 issue of American Pharmaceutical Review - Volume 14, Issue 6. Copyright rests with the publisher. For more information about American Pharmaceutical Review and to read similar articles, visit www.americanpharmaceuticalreview.com and subscribe for free.