Introduction
What is the solubility of my compound? During research and development phases for drugs, the question is frequently asked, “What is the solubility of my compound?” While at a first glance the answer seems to be just a simple number, it turns out that a useful answer will be much more complicated for several reasons. Factors which make answering the question complicated include the specific reason why solubility is important, certain properties of the compound itself as well as technical limitations in methods used to determine solubility.
Reasons for Determining Solubility
Solubility represents an important parameter during pharmaceutical research and development. Nevertheless, solubility is not a parameter which finally matters for a drug on the market. A drug does not have to be highly soluble, but the formulation of a drug has to provide “suitable” bioavailability. In this context, “suitable” means high enough and sufficiently reproducible to make a drug efficient and safe. Accordingly, the question of, “What is the solubility of my compound?” translates into the pharmacokinetics, pharmacodynamics and toxicodynamics of the compound. In comparison to bioavailability, solubility offers the big advantage of representing a property which can be easily measured.
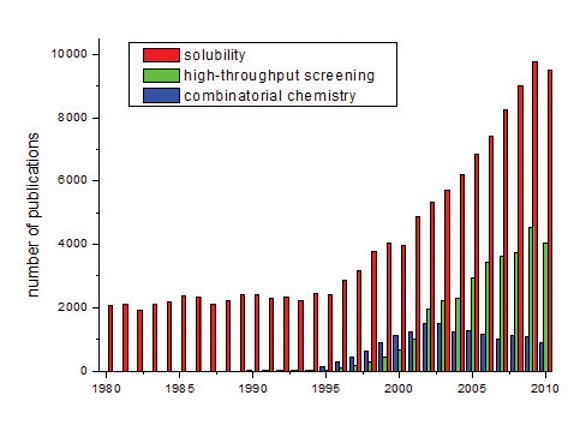
Figure 1- Evolution of numbers of publications on solubility, highthroughput screening and combinatorial chemistry over the last two decades. Increasing interest in solubility - reflected in numbers of publications – started almost parallel with the advent of highthroughput screening and combinatorial chemistry.
The number of publications dealing with solubility has been increasing continuously (Figure 1). In this context, the scientific community is not only struggling with solubility in the pharmaceutical world but also in other disciplines where challenges are quite similar, e.g. plant protection or pest management. Also, we have seen a considerable change in the solubility of pharmaceutical research and development compounds as well as of drugs which have been approved during recent years, indicating a clear trend towards lower solubility [1,2]. This trend is also reflected if allegorized in the percentage of compounds falling into the different categories in the biopharmaceutical classification schemes (BCS) [3]. There, clearly a trend towards higher percentages falling into BCS class II (high permeability, low solubility) and IV (low permeability, low solubility) is observed. As the BCS scheme does not only take into account solubility but also the dose of a drug and, accordingly, the BCS classification depends on potency, we can see that the trend in the solubility of pharmaceutical research and development compounds towards lower solubility is not compensated by a trend towards improved potency. Reasons behind this trend have frequently been discussed in terms of the influence of combinatorial chemistry and high-throughput screening (HTS) but also the selection of drug targets has an important influence on the solubility of compounds dealt with in a research and development program.
These aspects – especially the technical changes in pharmaceutical research and the solubility of the research and development compounds – have put solubility in a new context. Whereas during the early days of pharmaceutical research compounds moved on quite quickly from synthesis to animal trials, today we have many assays in between which deliver information about the compounds in a stepwise fashion [4,5]. Typically, the cascade starts by assessing the activity of a research compound in a biochemical or cellular assay including the target and interference assays. If the outcome of these assays is positive, screening will be continued by e.g. examining plasma stability, microsomal stability, lipophilicity, plasma protein binding and permeability. In addition, toxicological parameters such as genotoxicity or hERG binding will be assessed also by in-vitro assays. For all of these assays, the important pre-condition is sufficient solubility of the research compound in the respective medium used in the assay. The variability of the media used in the diverse set of assays is considerable. At the same time, a large number of research compounds are profiled. This holds especially true if high-throughput screens using biochemical and cellular assays are considered. Consequently, it becomes obvious that, even if solubility clearly represents a key parameter for these assays, compromises have to be made at least with regards to the selection the solvent used for determination of solubility. Thus, in most cases a generic solvent will be used to assess solubility as a starting point for the early research phase. In addition, it has to be kept in mind that in most cases the assays named above will be carried out using the pre-dissolved compound, e.g. 10 mmol/L DMSO solutions which are pipetted into the medium used in the assay (kinetic solubility). Therefore the question, “What is the solubility of my compound?” will translate into, “Which concentration will be obtained if the compound precipitates?” It is important to distinguish this question from the question, “To what extent does the compound dissolve?” as it is introduced into a solvent as a solid compound (thermodynamic solubility). Generally kinetic solubility will be higher or equal to thermodynamic solubility, as oversaturated solutions are easy to obtain by using kinetic conditions – the pre-dissolved compound – and are much harder to obtain under thermodynamic conditions using the solid compound [6].
As a research program moves on and interest focuses on a decreasing number of compounds synthesized during lead finding and lead optimization, answering the question, “What is the solubility of my compound?” will take into account more specific parameters. These include the properties of the respective assay buffers, e.g. pH of the medium or co-solvents which might be present and in which the compounds generally are introduced from a DMSO stock solution. Nevertheless, at this stage solubility is still required to ensure that the compound does not precipitate at the concentration used in the assay, and thus does not generate false-negative or false-positive results due to undissolved compound in the assay format [7].
Moving still further on with a research program puts an eye on bioavailability of the compound – the crucial parameter. Now the question, “What is the solubility of my compound?” translates into, “To what extent does my compound dissolve?” Consequently, this has to be answered by measuring thermodynamic solubility using the solid compound. At this stage already a formulation used for clinical trials and the marketing formulation should be borne in mind. The simplest and most cost-effective approach for these formulations in many cases will be development of an oral solid dosage form, e.g. a tablet or a powder-filled capsule. Nevertheless, before clinical trials are started, profiling of the compound in animal experiments such as pharmacokinetics, pharmacodynamics and toxicology has to be carried out. These experiments will be done using either the compound which is dissolved in a suitable vehicle, e.g. a system containing a co-solvent, a surfactant or cyclodextrines. Alternatively, if the oral route is used, the undissolved compound can be administered, e.g. as suspension or directly as powder material. In these cases dissolution has to take place in-vivo and probably the question, “What is the solubility of my compound?” translates into the most complex language. Assessing solubility – and also dissolution – under in-vivo conditions has to take into account different parts of the gastrointestinal tract (e.g. stomach or different parts of the intestine) and physiology in different species (e.g. mouse, rat, dog, monkey, human) under different conditions (e.g. fasted state, fed state, co-administration of other drugs such as protonpump inhibitors or antacids) [8]. It becomes obvious at this stage at the latest that the number of parameters influencing solubility becomes enormous. To render the situation still more complicated, not only the composition and properties of the solvent have to be considered but also the behavior of the compound itself. These include principal properties of the compound such as its protolytic behavior (pKa values) which will effect formation of salt forms as well as solid state properties such as formation of polymorphs or pseudo-polymorphs, e.g. hydrates. Further on these parameters will have a double impact on a research and development project: they will be relevant to the in-vivo behavior of a compound but can also be used by medicinal chemists to improve the properties of a compound e.g. by selection of pharmaceutical salts [9] and selection of the final solid state form for formulation development [10]. At the extremes this could be the thermodynamically most stable polymorph which will exhibit the greatest stability going hand in hand with lowest solubility or an amorphous or pre-dissolved formulation which will provide access to improved bioavailability.
Methods of Determining Solubility
Depending on the context of the question, different assays including analytical methods are used to answer the question, “What is the solubility of my compound?” Usually the first time the question is asked for a specific compound is during the design stage of the compound by the chemist. Typically at this stage different methods can be used for insilico assessment of solubility [11].

Figure 2-Modular setup of assays for solubility determination. Methods to determine solubility are organized in four consecutive steps. According to the specific needs these steps can be combined to yield a customized assay for determination of solubility.
After the compound has been synthesized by a medicinal chemist, usually determination of kinetic solubility is measured for a vast number of compounds, e.g. hits from HTS runs or compounds which are synthesized during optimization within a chemical series. Methods used for this purpose have to fulfill requirements such as providing high throughput including automation of the measurement, requiring only a minimal amount of the compound and providing sufficient sensitivity. The first three of these requirements are usually addressed by introducing the research compound into the assay format as a solution in a standard format, e.g. 10 mmol/L solution in DMSO which is provided in a standard format such as 40 μL vials. This allows easy handling of the compounds as pipetting can be done either manually or using a robot. Generally, methods for determining solubility can be separated into methods which do or do not require a phase separation of the solution and the undissolved residue (Figure 2). The latter methods are mainly represented by nephelometric or turbidimetric assays to determine kinetic solubility [12]. In these assays a dilution series of the compound is prepared in a well-plate by pipetting the compound into an aqueous buffer system and sequential dilution steps are carried out. Upon introduction of the compound into the first well containing the aqueous buffer, the compound precipitates rendering a turbid suspension. Starting from this suspension, several dilution steps are carried out by pipetting a defined fraction from this suspension to the next well of the well plate, generating a suspension with a lower concentration of the compound. As long as the concentration in the suspension is higher than the solubility of the compound, there will be a precipitate. As soon as the concentration in one well becomes lower than the solubility, the compound will completely dissolve in the buffer and a clear solution will be obtained. The readout is done using a nephelometer which measures the turbidity of all the wells belonging to one dilution row. Clearly, the advantages of this nephelometric assay are the speed of the assay, as readout by nephelometry is done within seconds, and ease of handling because only pipetting steps are required. Nevertheless, there are also critical parameters for this method. Probably the most important one is represented by the sensitivity of the method: typically, nephelometric solubility allows determination of kinetic solubility down to about 20 mmol/L [7]. Below this concentration in many cases the assay cannot differentiate solubility. This is due to the fact that for lower concentrations the content of a well will appear clear – without turbidity – to the eye of the nephelometer, as the compound might either be completely dissolved or not. If the compound is not dissolved, e.g. at a concentration of 5 μmol/L, the amount of precipitate will be to low to generate an amount of turbidity which can be recognized by the nephelometer. Therefore measuring solubility below 20 μmol/L typically is not feasible by nephelometry. This low sensitivity has to be especially taken into account if solubility should be determined to answer the question, “What is the solubility of my compound?” in order to decide whether the result of an assays which is carried out e.g. at a concentration of 1 μmol/L is reliable or not.
Methods to determine kinetic solubility with higher sensitivity require phase separation between the liquid phase containing the dissolved compound and the undissolved solid residue [13]. Following this phase separation, determination of the concentration in the saturated solution is carried out by a suitable analytical technique. These two steps represent the basic steps for a setup of assays to determine kinetic solubility which can be customized according to specific needs (Figure 2). The first crucial step – phase separation – might be carried out either by centrifugation or by filtration. The second crucial step – determination of concentration in the saturated solution after incubation of the suspension for a welldefined period of time at a well-defined temperature – can be carried out by different analytical techniques such as UV-Vis spectroscopy [14], HPLC-UV [15, 16], HPLC-MS [17] or even other methods such as HPLCELSD (Evaporation-Light-Scattering-Detector) or HLPC-CNLD (Chemical- Nitrogen-Luminescence-Detector). In principle, any combination of these two key steps can be combined to yield an assay format for determination of kinetic solubility. Nevertheless, several considerations have to be taken into account when setting up a specific assay and when results of the assay are considered. For the first step usually phase separation by centrifugation overestimates solubility as, after centrifugation, small particles might still be present in the supernatant which will be dissolved after the phase separation either by adding a co-solvent to avoid further precipitation of the saturated solution before analysis or by the HPLC eluent. On the other hand, using filtration of the suspension for phase separation might underestimate the solubility of the compound as it might adsorb to the material of the filter.
The second crucial step of the solubility determination represented by an analytical technique to determine the concentration of the compound in the saturated solution also requires careful choice of the method. Whereas UV spectroscopy, like nephelometry, clearly offers the advantage of high speed, limitations are also sensitivity of the method and the inability of the method to detect impurities to degradation products. Both disadvantages are avoided if HPLC coupled with a suitable detection is used for determination of the concentration in the saturated solution. Clearly, degradation products and impurities can be detected and effects from these can be separated from effects from the research compound itself. Furthermore even if HPLC is just used in combination with UV-detection, sensitivity down to 1 μmol/L is easily realized. The sensitivity can be further lowered if HPLC is used in combination with mass spectroscopy down to 0.1 μmol/L or still even lower. On the other hand, using HPLC has the disadvantage of requiring a longer time for analysis of one compound and, especially in the case of HPLC-MS, more effort to adjust the method to a special research compound. The first disadvantage is at least partly avoided by the advent of UPLC techniques allowing faster gradients. All of these coupled methods, however, require a considerable higher investment.
As soon as the question, “What is the solubility of my compound?” is asked in the context of thermodynamic solubility, the answer can be provided by assay formats which are very much the same as those described to determine kinetic solubility and which rely on phase separation of the solid and liquid phase. The main difference is just given by the fact that the compound is introduced as solid material into the aqueous buffers system instead of using the compound pre-dissolved in DMSO. This approach also avoids effects of DMSO which acts as a co-solvent in kinetic solubility assays. Most importantly, the price to pay for determining thermodynamic solubility is based on the more time-consuming handling of the compound in solid state, which usually cannot be automated as research compounds frequently are sticky, oily, glassy or electrostatic. Additionally, compared to the amount of compound which is required for determining kinetic solubility (e.g. 30 μL of a 10 mmol/L DMSO solution), the amount of compound required to determine thermodynamic solubility is higher. Nevertheless, even in these assays formats it is possible to get meaningful results with just 2-3 mg of the respective compound.
As an important parameter also the solid state properties of a compound can be taken into account in the thermodynamic solubility assay. Solubility is defined as the equilibrium state between the solid residue and the saturated solution which is in contact with the solid residue. Consequently, different results for solubility will be obtained if the solid residue is represented by a crystalline phase or by amorphous material. Bearing in mind formulations based on the solid active pharmaceutical ingredient, usually a crystalline phase will be the target of the research and development program. Consequently, it has to be guaranteed that thermodynamic solubility results also refer to a crystalline phase [6]. This question can be answered by investigating the solid state residue obtained after incubation of the suspension by techniques such as polarized-light microscopy or powder X-ray diffraction (PXRD). Whereas the first one offers advantages such as being less labor-intensive and requiring only a minimal amount of the compound, PXRD allows more in-depth understanding of the actual phase which is obtained as solid state residue. Understanding of the solid state residue can be still improved as regards presence of salts by ion chromatography (IC). This topic becomes especially important as soon as the question, “What is the solubility of my compound?” is answered as described above in the most complicated context of dependency of various parameters such as pH, variable concentrations of ions being present in the respective media including simulation of biorelevant media [18]. In this environment where a more in-depth understanding of solubility and parameters influencing solubility is required, analytical characterization of the solid state residue obtained after incubation of the compound e.g. by PXRD and IC becomes a crucial step beyond determination of the concentration of a compound in solution.
Summary
During pharmaceutical research and development, solubility of compounds has to be assessed according to the special need of the respective situation. Over the periods of early research phases, kinetic solubility of a large number of compounds has to be measured under more generic conditions. Later on in research and development the answer of the question, “What is the solubility of my compound?” is given only for a lower number of compounds. At this stage solubility is assessed more in depth, taking into account different conditions by measuring thermodynamic solubility. For all purposes mentioned above, methods to determine solubility can be customized individually from a toolbox containing mainly four tools which can be combined as modules: Introduction of the compound – Equilibrating the liquid and the solid phase – Phase separation and Determination of the compound concentration in the liquid phase.
References
- C. Lipinski, „Poor Aqueous Solubility – an Industry wide Problem in Drug Discovery“, Am. Pharm. Rev. 2002, 5, 82-85.
- M.C. Wenlock, R.P. Austin, P. Barton, A.M. Davis, P.D. Leeson, „A Comparison of Physicochemical Property Profiles of Development and marketed Oral Drugs“, J. Med. Chem. 2003, 46, 1250-1256.
- Amidon G.L., Lennernäs H., Shah V.P., Crison J.R. „A Theoretical Basis for a Biopharmaceutics Drug Classification: The Correlation of In Vitro Drug Product Dissolution and In Vivo Bioavailability.“, Pharm. Res. 1995, 12, 413-420
- E.H. Kerns, L. Di, „Drug-like Properties: Concepts, Structure Design and Methods – From ADME to Toxicity Optimization“, Academic Press 2008, London.
- B. Faller, L. Urban, „Hit and Lead Profiling“, Wiley-VCH 2009, Weinheim.
- C. Saal, „Optimizing the Solubility of Research Compounds: How to Avoid Going Off Track“, Am. Pharm. Rev. 2010, 13, 14-18. 7.
- B. Hoelke, S. Gieringer, M. Arlt, C. Saal, „Comparison of Nephelometric, UV-Spectroscopic, and HPLC Methods for High-Throughput Determination of Aqueous Drug Solubility in Microtiter Plates“, Anal. Chem. 2009, 81, 3165-3172.
- T.T. Kararli, „Comparison of the Gastrointestinal Anatomy, Physiology, and Biochemistry of Humans, and commonly used Laboratory Animals“, Biopharm. Drug. Disp. 1995, 16, 351-380.
- P.H. Stahl, C.G. Wermuth, „Handbook of Pharmaceutical Salts: Properties, Selection and Use“, Wiley-VCH, 2011.
- R. Hilfiker, „Polymorphism in the Pharmaceutical Industry“ Wiley-VCH 2006, Weinheim.
- J.C. Dearden, „In silico Prediction of aqueous Solubility“, Expert Opin. Drug Discov. 2006, 1, 31-52.
- C.D. Bevan, R.S. Lloyd, „A High-Throughput Screening Method for the Determination of Aqueous Drug Solubility using Laser Nephelometry in Microtiter Plates“, Anal. Chem. 2000, 72, 1781-1787.
- D. Roy, F. Ducher, A. Laumain, J.Y. Legendre, „Determination of the Aqueous Solubility of Drugs Using a Convenient 96-Well Plate-Based Assay“, Drug Dev. Ind Pharm. 2001, 27, 107-109.
- T.-M. Chen, H. Shen, C. Zhu, „Evaluation of a Method for High-Throughput Solubility Determination using a Multi-wavelength UV Plate Reader“, Comb. Chem. High Through. Screen. 2002, 5, 575-581.
- E.H. Kerns, „High-throughput Physicochemical Profiling for Drug Discovery“, J. Pharm. Sci. 2001, 90, 1838-1858. 1
- A. Glomme, J. Maerz, J. B. Dressman, „Comparison of miniaturized Shake-Flask Solubility Method with automated potentiometric acid/base Titrations and calculated Solubilities“, J. Pharm. Sci. 2005, 94, 1-16.
- Z.B. Alfassi, R.E. Huie, B. L. Milman, P. Neta, „Electrospray Ionization Mass Spectroscopy of Ionic Liquids and Determination of their Solubility in Water“, Anal. Bioanal. Chem. 2003, 377, 159-164.
- M. Vertzoni, A. Diakidou, M. Chatzilias, E. Soederlind, B. Abrahamsson, J.B. Dressman, C. Reppas, „Biorelevant Media to simulate Fluids in the Ascending Colon of Humans and their Usefulness in Predicting Intracolonic Drug Solubility“, Pharm. Res. 2010, 27, 2187-2196.
Author Biographies
Christoph Saal studied chemistry at the Technical University of Darmstadt and graduated in Physical Chemistry. In 1999, he joined Merck KGaA where worked in Central Analytics and Medicinal Chemistry. Currently, Christoph Saal is heading a group focused on analytical and physico-chemical characterization of New Chemical Entities. This field of activity includes solid state characterization as physico-chemical charaterization of pharmaceutical salts and polymorphs.
Anna Christine Petereit has studied Pharmacy at the Goethe University of Frankfurt and she will briefly finish her PhD in Pharmaceutical Technology targeting the prediction of oral bioavailability based on surface activity profiling. She has further experience in formulating solid oral dosage forms. Currently Anna works as a Scientist at Merck KGaA in the division for physical-chemistry properties characterisation with the main focus on solubility and dissolution.
This article was printed in the July/August 2011 issue of American Pharmaceutical Review - Volume 14, Issue 5. Copyright rests with the publisher. For more information about American Pharmaceutical Review and to read similar articles, visit www.americanpharmaceuticalreview.com and subscribe for free.