Introduction
Since the beginning of development of monoclonal antibodies (mAb) in the 1980s, they become vastly successful as efficacious and safe therapeutics for a variety of diseases. Today, they represent one of the fastest growing segments in the pharmaceutical industry.
Nevertheless, the resources and risks associated with the development of new antibodies are immense and reflected in the low number of only ~ 33 antibodies that have been approved in the US whereas 4 were later withdrawn by the sponsor [1].
Historically, the pharmaceutical development of antibodies was a customized approach for each new molecule that proceeded from Discovery to Development. The efforts were predominantly focused to establish a tightly fixed process and to assure quality by compliance. The growing perception that this limits a continuous improvement and the use of new technologies to further increase safety and efficacy of biologics, lead to new development concepts trying to accommodate both, the business needs to optimize efficiency in development and the expectations by authorities towards an increasing drug product quality by enhancing product and process understanding, as outlined by the concept of Quality-by-Design (QbD) [2].
With ~ 240 mAbs currently in clinical trials, companies who have a large number of antibodies in mid- and late-stage discovery are looking for new strategies to enable a “routine early development,” based on their extensive experience gained with previously developed molecules.
Following the concept of QbD, Platform Technologies as stated in the A-Mab Case Study are considered a valuable tool to improve the efficiency and quality in drug product development [3]. The basic idea is that a Development Platform, in combination with a risk-based approach, is the most systematic method to leverage Prior Knowledge for a given new molecule. Furthermore, such a platform enables a continuous improvement by adding data for every new molecule developed by this approach, hence increasing the robustness of the platform.
The concept of how to establish and apply such a Technology Platform for a given process is still a relatively new approach and up to now there is no definition available by official guidance documents on the pharmaceutical development like the ICH Q8(R2).
However, following the concept of QbD, a Technology Platform can be defined as:
A systematic approach to leverage Prior Knowledge for standardized processes that have been demonstrated that the multidimensional combination and interaction of input variables and process parameters can be applied for a class of molecules with comparable characteristics to provide assurance of quality.
As outlined in this definition, a key prerequisite to establish and apply a technology platform is the possibility to identify a class of molecules showing comparable characteristics in terms of their physico-chemical properties and stability profile, like monoclonal antibodies. Every molecule that matches these characteristics can be treated as a nextin- class molecule.
Processing these molecules by using standardized equipment and manufacturing parameters can be described by one Platform Design Space assuring a comparable quality of the final drug product.
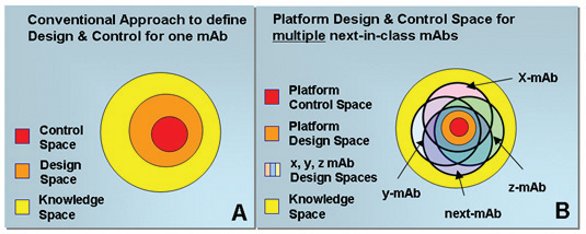
Figure 1. Illustration of a conventional Knowledge, Design and Control Space A) and the adopted concept for a Technology Platform (B)
Figure 1A illustrates the concept of a Knowledge, Design and Control Space as it refers to a single molecule. How to adopt the very same concept for multiple next-in-class molecules is captured in Figure 1B. Based on the available Knowledge Space for x, y & z-mAb a Design Space can defined for each of the molecules. Although there might be slight differences between the individual Design Spaces, they overlap to a great extent. This overlap allows defining a Platform Design Space that can be significantly smaller compared to the individual ones, but is still broad enough to match the process capabilities. Due to the strict application of standardized formulations, equipment and unit operations in this early development stage, the additional flexibility of a broad Design and Control Space is not required. If a new molecule (next-mAb) has comparable characteristics (e.g., stability profile) to those used to establish the platform (x, y, z-mAb), it can be considered a next-in-class-molecule covered by the established Platform Design & Control Space.
Prerequisites to Establish a Technology Platform for Early Development
One of the key elements in establishing and justifying the applicability of a Technology Platform is to identify those molecules that match the definition of a next-in-class molecule.
A systematic approach to assure that a selected lead molecule candidate proceeding into development is a next-in-class molecule, requires a combined effort of multiple functions in late-stage discovery, involved in candidate generation, screening and selection of the lead candidate.
Target Molecule Profile
imilar to the definition of a Target Product Profile to guide the development efforts for a drug product, the respective approach to guide the candidate selection process will start with the definition of a Target Molecule Profile (TMP), specifying the key characteristics required for a molecule to assure stability, and hence, safety and efficacy.
Table 1. Example of a Target Molecular Profile (TMP) for monoclonal antibodies
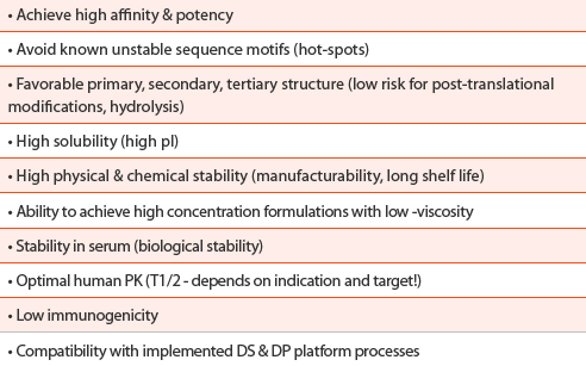
Therefore, a standardized TMP as outlined in Table 1 is not only focusing on the biologic function, like affinity or potency, but equally weights the drug-like properties of the molecule to assure broad formulation options needed to provide a competitive product to the market (e.g., a high-concentration liquid formulation). Finally, the standardized drug-like properties specifications guide the selection towards comparability to previously developed mAbs matching the same TMP. Therefore, with regards to QbD, the TMP is the starting point and of paramount importance to design quality into the molecule, thus into the final drug product.

Figure 2. Flow-chart of a standardized screening process to assess and identify next-in-class molecules as defined by the Target Molecular Profile (TMP)
Candidate Screening & Selection
Based on the TMP all potential molecule candidates will be submitted to a standardized screening process as presented in the flow-diagram in Figure 2 to assess their respective drug-like properties and to assure that the selected lead candidate molecule matches the next-in-class criteria, justifying the application of the platform process. This screening process again involves several functions and is carried out in multiple tiers, adapted to the number of candidates to be screened at each tier. The first tier assay is usually focused on the primary amino acid sequence and in-silico screening methods to identify sequence and structural liabilities that might cause chemical or physical degradation (deamidation, aggregation, colloidal stability) or are associated with unwanted biological effects like immunogenicity.
Since the accuracy of current liability screening methods does not allow ruling out candidates solely on in-silico methods, subsequent testing will provide the necessary data to finally assign a development risk to each and every molecule. This allows a ranking according to predefined benchmark criteria based on Prior Knowledge. Only those molecules that showed the overall best drug-like properties, and hence, have the lowest development risk will be selected as lead and backup candidates.
Finally, the outcome of the screening allows justifying whether the selected lead and backup candidate is a next-in-class molecule, and hence, the application of the platform process.
If none of the screened molecules will match with the requirements of a next-in-class molecule, more extensive and customized formulation and process development efforts are needed.
Establishing a Platform Process in Early Drug Product Development
All the previously described activities will take place in late-stage discovery and will be directed towards enabling a platform process in development to efficiently provide clinical supply for phase 1 trials.
The following section gives an overview about the activities that need to take place in the development area.
Since general outlines of using QbD in pharmaceutical development for biologics are described in details elsewhere[3], the focus will lay on the specific activities around establishing and applying a technology platform.

Figure 3. Flow-chart of the various steps to implement a Technology Platform
As visualized in Figure 3, the methodology to set up the platform closely follows the concept of QbD. Still the systematic approach starts with defining a standard target process, which will be used for all next-inclass molecules.
What follows is the identification of all process parameters e.g., via a fishbone diagram that will be subsequently analyzed using a risk-based approach on their impact on the Quality Attributes of the molecule. Given the early development stage and the lack of clinical experience, the definition of critical Quality Attributes relies on the available in-vivo data and Prior Knowledge (previously developed mAbs, guidelines, etc.).
The subsequent efforts of exploring the Knowledge Space depend on the available Prior Knowledge, especially during the implementation of the platform process. These activities would require additional experiments to gain the increased product and process understanding necessary to define a Design and Control Space. Once the Platform Process is established and has been successfully demonstrated for multiple molecules, no additional formulation or process development activities are needed. The information gained in late discovery platform screening will justify that the lead molecule can be considered a nextin- class molecule and falls into the Design & Control Space of the established platform.

Figure 4. Flow-chart of the various steps to implement a Technology Platform
An example of how a platform development approach can be implemented as a part of the development activities is outlined in Figure 4. Starting with limited Prior Knowledge for the given class of molecules, initial activities would focus on the basic characterization and formulation and process development. Extensive initial screening studies will provide information on suitable formulations. Ideally, a standardized manufacturing process will be applied focusing on robustness rather than efficiency to simplify process transfer activity. As an outcome of the stability assessment a lead formulation will be selected to be used for subsequent clinical supply manufacturing. Furthermore additional backup formulations will be selected to be tested with any new molecule of the same class.
The next phase is designed to verify whether the previously identified formulation can be used for the molecules y- and z-mAb of the same class exhibiting comparable characteristics (next-in-class molecules).
Both molecules will be extensively characterized and their stability in the lead as well as in the backup formulations will be analyzed. If the data for y- and z-mAb show comparable stability to x-mAb, the standardized formulation and manufacturing can be used as a platform for this class of molecules. Due to the extensive preformulation candidate screening and the application of Prior Knowledge from x-, y- and z-mAb, any following next-in-class molecule like next-mAb can be directly submitted for clinical supply manufacturing. Subsequent IND stability studies will finally verify the use of the platform process.
Conclusion
Modern advancements in protein engineering give the tools to identify and generate a large number of potential antibody candidates towards a given target. Using a systematic screening approach allows the assessment of their drug-like properties and to identify the most promising molecules that match predefined stability criteria, as specified in the target molecule profile. All molecules that match with these criteria would show comparable characteristics and become next-in-class molecules, which can be developed by using a technology platform using a standardized formulation e.g., composition, excipients, and packaging components as well as standardized process equipment and unit operations.
Such a systematic approach to use Prior Knowledge can significantly reduce the development efforts needed to initiate phase I clinical trials, while continuous improvment of the product and process understanding with every molecule of the respective class. Finally, an additional benefit is the increased predictability and accuracy of development timelines allowing sychronization of multiple projects more efficiently.
References
- J.M. Reichert, Metrics for antibody therapeutics development, Mabs (2010), 2(6): 695-700.
- ICH Harmonized Tripartite Guideline: Pharmaceutical Development Q8(R2). Step 4 version, Aug. 2009.
- CMC Bio Working Group 2009. A-MAb: A Case Study in Bioprocess Development. Version 2.1, California Separation Science Society.
Author Biography
Michael Siedler joined Abbott GmbH & Co KG in 2007. Prior to that, he worked at Scil Technology GmbH in Martinsried, Germany. He received his Ph.D. in chemistry from the University of Hamburg in 2002.
This article was printed in the September/October 2011 issue of American Pharmaceutical Review - Volume 14, Issue 6. Copyright rests with the publisher. For more information about American Pharmaceutical Review and to read similar articles, visit www.americanpharmaceuticalreview.com and subscribe for free.