Abstract
Raman scattering technology has been found to be a very useful tool to aid in the development of well-understood solid dosage forms with appropriate manufacturing controls and storage conditions. With peaks related to fundamental vibrational modes, Raman spectroscopic data can be used to analyze the solid-state behaviors of active pharmaceutical ingredients (APIs) in drug products such as polymorphic conversion, formation of amorphous material or crystallization, and changes in ionization status caused by excipient interactions on exposure to varying conditions of heat or humidity. In this article, applications of Raman scattering technology to explore the solid-state properties of API in tablet formulations are discussed with case studies.
Introduction
Control of the solid-state form of the active pharmaceutical ingredient (API) in formulations can be a critical quality attribute affecting the safety, performance and efficacy of the resulting drug products. Tablets, as the most commonly used solid dosage forms, present a complicated situation for analysis of these solid state forms due to dilution effects and interference from excipients. It is challenging both from the sensitivity and specificity standpoints to monitor API form conversions when dealing with different combinations of excipients, but the information can be critical to guide the formulation optimization process and to establish consistency of the solid-state form during manufacturing and stability.
Good characterization and control of the drug substance and drug product manufacturing processes are key to ensure the drug product quality. Testing of drug substance alone is often performed from different perspectives including structural composition, physical properties, chemical purity, and chemical stability. One of the best scenarios is to keep the API in the tablet in the same solid-state form as the drug substance at release and during shelf life. However, there are many cases in which the solid-state form of drug substance undergoes changes including amorphous formation, polymorph conversion, or ionization status change during the drug product manufacturing process and storage due to mechanical force, heat, moisture, and excipient matrix effects [1, 2]. These form conversions can potentially lead to undesired product appearance, loss of potency, and even changes in clinicalexposure, and therefore their rates and mechanisms need to be studied using techniques with the requisite sensitivity and specificity.
Numerous methods have been used to characterize the solid-state forms of API in tablet formulations, including X-ray diffraction (XRD), thermal analysis (differential scanning calorimetry (DSC)), solid-state nuclear magnetic resonance (ssNMR) spectroscopy, vibrational spectroscopy (including infrared(IR), near infrared (NIR), and Raman), and optical microscopy. Technologies such as ssNMR or XRPD are sometimes preferable since they have relatively obvious data interpretation with non-overlapping sharp peaks that can be directly assigned to diff erent solid-state forms, while Raman spectroscopic data often require more sophisticated analyses including applications of appropriate chemometric algorithms.
With the advantages of sampling flexibility, large Raman scattering cross-sections of aromatic APIs, and facile instrumentation operation and maintenance, Raman scattering technology is often found to be the viable choice among complementary technologies. This was found to be the case especially when analyzing amorphous API content present at low levels in a complex sample matrix by XRPD [3], or for determination of API in an interfering excipient matrix by 1H-ssNMR.
The study here describes the use of Raman spectroscopy to analyze the critical properties of APIs in formulations containing excipients that are commonly used in tablets.
Experimental
Raman Measurements
The Raman spectra of samples from the various drug substances were measured using a Raman microscope system for solid samples, and a Raman analyzer for solution samples. The excitation source for the Raman microscope system is 532 nm laser of ca. 300 mW at the samples through a 5 x objective with a sampling area of about 450 μm in diameter. A typical measurement of a formulated solid sample may require at least 30 minutes of photobleaching time prior to the Raman signal acquisition. The Raman analyzer has a probe which uses a 500 mW 785 nm laser excitation.

Figure 1 - Raman Spectra of 10 mg DSI Tablet after 6 months storage at (a) 40°C/75%RH and (b) 25°C/60%RH, and the reference Raman spectra of (c) placebo tablet, (d) amorphous DSI, and (e) crystalline DSI.
Results and Discussion
Amorphous API Content in Tablets
The relative amounts of amorphous and crystalline API in tablets play a critical role in the quality of drug product from both the chemical and physical perspectives. Due to processability considerations and superior purity and stability, the crystalline form of the API is often preferred for drug substance when suffi cient clinical exposure can be achieved for drug product. The conversion of crystalline API to the amorphous state in the presence of some excipients especially under accelerated storage conditions with high temperature and humidity are well-known in the literature [4-6]. In general, the amorphous state is known to often have higher rates of degradation, and superior dissolution as compared to the crystalline form. These changes can potentially affect the potency, safety and clinical exposure. In addition to the HPLC assays and dissolution tests which are often used to evaluate the potency, purity, and dissolution profi les of tablets, the direct measurement and quantitation of the amorphous content can be a critical quality attribute in achieving rational formulation development and the choice of appropriate storage conditions utilizing quality-by-design considerations.
Drug substance I (DSI) is a free acid crystalline hydrate with dehydration temperature at about 60° C. The solid dosage form of DSI is a 10 mg tablet with 5.3% drug load. As a BCS class II compound, it was requested to monitor amorphous content after tablet manufacturing and stability. Based on the Raman measurements of the neat crystalline DSI and its amorphous form, the aromatic carbon double bond stretching region at around 1610 cm-1 was found to be the most sensitive spectral region for the determination of amorphous API content in the tablet, as shown in Figure 1. Since the common excipients used in tablet formulations are predominantly aliphatic molecules, the aromatic portion of the DSI molecule is the exclusive contributor to the spectral region at 1500 - 1700 cm-1. The increased amorphous content in the tablets can be identifi ed by checking the relative peak intensities, and the quantifi cation of the amorphous content can be determined by using multivariate algorithms as described elsewhere [6].
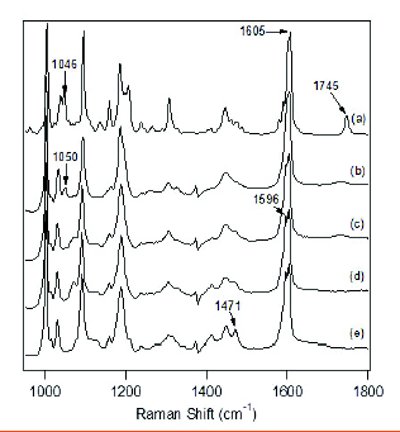
Figure 2 - Raman spectrum of (a) crystalline DSI, and solution Raman spectra of DSII with diff erent equivalents of (b) 2 HCl, (c) 1 HCl, (d) 0 HCl, and (e) 1 NaOH with 20 mg/mL DSI in 20/80 acetonitrile/water (v/v).
Evidence of API Deprotonation in Formulation with Excipients
Another phenomenon that can alter the chemical stability of an API relates to its ionization status change in tablets after manufacturing and storage. In this case, even though the drug substance itself showed suffi cient chemical stability through accelerated storage conditions, the formulated tablet still exhibited problematic chemical stability with possible excipient-related causes. One of the common causes of poor chemical stability of API in tablets has been previously attributed to the ionization status change of the API in the pH microenvironment formed by various excipients [2, 4, 7]. A study using thermodynamic concepts for determination of the rates of a deprotonation reaction in the solid- state between API and excipients has been reported in the literature [8]. Direct characterization of solid-state acid-base reactions with technologies including 13C-ssNMR, mid-IR, and Raman spectroscopy also has been published [1, 9].
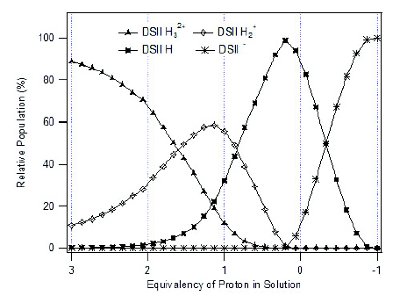
Figure 3 - Positive or negative equivalent values stand for the number of HCl or NaOH equivalent in DSII solution.
Drug substance II (DSII) is a water soluble dihydrochloride (diHCl) salt with three pKa values of 2.1, 3.0, and 8.2, and aqueous solubility greater than 10 mg/mL. Since it is a salt formed of a weak base and a strong acid, the deprotonation reaction can be thermodynamically favored in neutral or relatively basic pH microenvironments existing in the excipient matrix [8]. For the purposes of tracking the deprotonation reaction of DSII especially in its amorphous state, the Raman spectra of DSII in solution with diff erent equivalents between HCl and DSII freebase and the crystalline solid of diHCl salt were measured, and are shown in Figure 2. The solvent spectral signal has been subtracted from all of the solution Raman spectra. Spectra (b)-(d) correspond to the equivalents of 2, 1 and 0 HCl to DSII freebase in solution, while Spectrum (e) was obtained from one equivalent of NaOH to DSII free base in solution. The peak at 1745 cm-1 can be used as an indicator for the existence of crystalline material, as the solution Raman spectrum is representative of its solid-state amorphous spectrum. It is also notable that the disappearance of the 1050 cm-1 peak, the splitting of the 1605 cm-1 peak, and the appearance of a peak at 1471 cm-1 can be used as characteristic API Raman features for the determination of its diff erent deprotonation stages. Figure 3 shows the relative population of all DSII related solution species at diff erent equivalents of HCl by calculation based on known pKa values and measured pH values through a titration process starting from 3 equivalents of HCl using NaOH solution. Each ionization form of DSII is confi rmed to be the major species in solution with the same stoichiometric ratio.
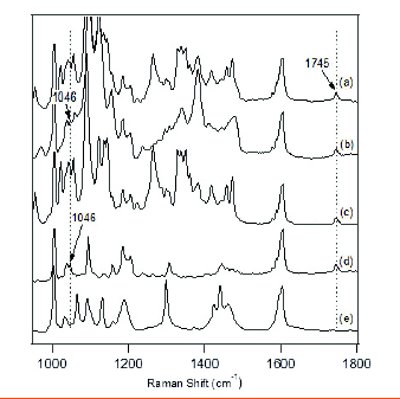
Figure 4 - Raman spectra of samples after 1 month storage at 40°C/75% RH for (a) DSII prototype formulation blend, and binary mixtures between (b) microcrystalline cellulose, (c) lactose, (d), silicon dioxide, (e) magnesium stearate, and DSII.
One prototype tablet formulation for DSII used 35% microcrystalline cellulose, 52% lactose, 1% silicon dioxide, 1% magnesium stearate, and 11% DSII by weight. The eff ect of the individual excipients was also checked by making binary mixtures with 10:1 ratio between microcrystalline cellulose or lactose and DSII, or 1:1 ratio between silicon dioxide or magnesium stearate and DSII. Figure 4 lists the Raman spectra of the drug blend and the four binary mixtures taken after one month storage at 40° C/75% RH. All samples except for the binary mixture with magnesium stearate show a clear crystalline peak at 1745 cm-1, which indicates that DSII remains in its crystalline form. The peak at 1046 cm-1 can also be identifi ed in both binary mixtures of DSII with microcrystalline cellulose and silicon dioxide. Again, the peak at 1046 cm-1 indicates the existence of the diHCl salt form, which is consistent with the observation that DSII remains in its original crystalline form with 2 equivalents of HCl. Even though the 1046 cm-1 peak in the Raman spectra of the lactose-containing samples is obscured by the lactose Raman bands in the same region, the presence of the crystalline peak at 1745 cm-1 still indicates some remaining crystalline API.
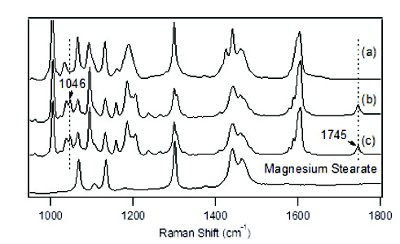
Figure 5 - Raman spectra of binary mixture between magnesium stearate and DSIIat the following conditions: (a) 1 month at 40°C/75%RH, (b) 40°C oven, and (c) initial sample.
Interestingly, the stressed binary mixture between DSII and magnesium stearate shows complete conversion of crystalline API to the amorphous state, with the disappearance of the 1745 cm-1 peak. The amorphous state of DSII also exhibits a deprotonation state with the disappearance of the 1046 cm-1 peak. Since no peak splitting is observed for the main peak located at 1605 cm-1 in fi gure 4(e), the Raman spectral change related to DSII in the binary mixture with magnesium stearate is consistent with the previous observation of DSII freebase with one equivalent of HCl, as shown in fi gure 2(c). It is concluded that DSII converts to its mono-acidic amorphous form in the sample matrix with magnesium stearate. To further investigate the possible cause of the humidity eff ect, Figure 5 compares Raman spectra of the same binary mixtures after one month storage at 40°C/75%RH and 40°C conditions, and also lists the reference Raman spectra taken from the non-stressed binary mixture sample and magnesium stearate. At 40°C without humidity, the solid-state form of DSII remains unchanged, which indicates that the combined eff ect of both humidity and magnesium stearate can lead to the amorphous conversion and deprotonation of DSII. Although the drug blend also contains magnesium stearate, the conversion may be inhibited in this case due to the dilution factor and to the low level of magnesium stearate used. A more systematic understanding can be achieved if quantitative conversion data is available, which is currently limited by signal-to-noise ratio due to fl uorescence background from samples when using the 532 nm excitation laser. Future studies with longer laser excitation wavelengths such as 1064 nm using FT-Raman instrumentation are planned.
Conclusions
Raman scattering technology is a powerful technique for the characterization and quantifi cation of the API solid-state form in tablet formulations. A control strategy with technologies capable of direct measurement of API solid-state forms in the drug product provides both a better understanding of its behavior and an effi cient way to ensure the design and development of rational formulations.
Acknowledgements
The authors thank the AR&D department at Amgen for the support.
References
- Rohrs, B.R., et al., Tablet dissolution aff ected by a moisture mediated solid-state interaction between drug and disintegrant. Pharmaceutical Research, 1999. 16(12): p. 1850-1856.
- Zannou, E.A., et al., Stabilization of the maleate salt of a basic drug by adjustment of microenvironmental pH in solid dosage form. International Journal of Pharmaceutics, 2007. 337(1-2): p. 210-218.
- Shah, B., V.K. Kakumanu, and A.K. Bansal, Analytical Techniques for Quantifi cation of Amorphous/Crystalline Phases in Pharmaceutical Solids. Journal of Pharmaceutical Sciences, 2006. 95(8): p. 1641-1665.
- Hailu, S.A. and R.H. Bogner, Eff ect of the pH grade of silicates on chemical stability of coground amorphous quinapril hydrochloride and its stabilization using pH-modifi ers. Journal of Pharmaceutical Sciences, 2009. 98(9): p. 3358-3372.
- Wardrop, J., et al., Infl uence of solid phase and formulation processing on stability of Abbott-232 tablet formulations. Journal of Pharmaceutical Sciences, 2006. 95(11): p. 2380- 2392.
- Xie, Y., et al., Quantitative Determination of Solid-State Forms of a Pharmaceutical Development Compound in Drug Substance and Tablets. International Journal of Pharmaceutics, 2008.
- Byrn, S.R., W. Xu, and A.W. Newman, Chemical reactivity in solid-state pharmaceuticals: formulation implications. Advanced Drug Delivery Reviews, 2001. 48(1): p. 115-136.
- Stephenson, G.A., A. Aburub, and T.A. Woods, Physical stability of salts of weak bases in the solid-state. Journal of Pharmaceutical Sciences, 2011. 100(5): p. 1607-1617.
- Guerrieri, P. and L.S. Taylor, Role of salt and excipient properties on disproportionation in the solid-state. Pharmaceutical Research, 2009. 26(8): p. 2015-2026.
Author Biographies
Yong Xie is currently a senior scientist in the Analytical R&D department at Amgen. He received his Ph.D. in Analytical Chemistry from Purdue University in 2005 and joined Amgen the same year. Since then he has been working in analytical development and concentrates on deliveries of small molecule drug substance and drug product with varieties of analytical technologies. His experience and interests also include chemometrics based vibrational spectroscopic method development for potential critical quality attributes involved in both solution and solid state processes.
Nina S. Cauchon is currently a director in the Analytical R&D Department at Amgen Inc (Thousand Oaks, CA). She received her Ph.D. from the School of Pharmacy at Purdue University. She worked at Warner-Lambert/ Parke-Davis (Morris Plains, NJ) and at Miravant Pharmaceuticals (Santa Barbara, CA) prior to joining Amgen. Her experience spans a broad range of analytical techniques (spectroscopy, chromatography), and pharmaceutics (solid-state characterization). In addition, she has led cross-functional CMC teams supporting product development and regulatory fi lings for molecules at various stages of clinical and commercial development