The Challenge of Formulation: Poorly Water Soluble Compounds and BCS Class II Compounds
The well-established successes in combinatorial chemistry and high throughput screening have resulted in the rapid identification of many highly potent lead pharmaceutical compounds with optimized pharmacodynamic properties, but relatively sub-optimal biopharmaceutical characteristics. A principle challenge is evident in the increasing trend towards the identification of lead compounds with lower aqueous solubilities, which is estimated to apply to be between 40-70% of all new drug candidates [1]. In recent years there has been some considerable change in emphasis aimed at directing efforts to ensure lead compounds have optimized biopharmaceutical properties, principally driven on the basis of the Biopharmaceutical Classification Scheme (BCS), which established a framework for classification drugs on the basis of the key biopharmaceutical challenges, namely solubility and permeability [2]. The BCS has been instrumental in guiding oral drug development to strive for lead compounds which will not be solubility and/or permeability rate limited. However, despite the great advances in understanding the key factors influencing poor oral absorption, there are indications that the number of drugs under development in BCS class II (i.e. low solubility high permeability) has actually increased. For example, it has estimated, by comparing marketed drugs (pre 2006) to pipeline drugs, that there has been an increase of BCS class II compounds from ~30 to 50-60% and a corresponding decrease in BCS class I compounds from ~40% to 10-20% [3]. In summary, the challenge to successfully develop poorly water soluble drug compounds is as great as ever and unlikely to change in the near to medium term.
Lipid-based Drug Delivery Systems (LBDDS)
Table 1. Selected examples of drugs, including commercial and in development drugs, where a LBDDS resulted in enhanced bioavailability.

A drug is classified as ‘poorly soluble’ when its dissolution rate is considered so slow that dissolution takes longer than the transit time past the prime absorptive region in the GIT [4]. When administered in conventional solid dose formulations, these compounds have a tendency to exhibit low bioavailability as their absorption is described as dissolution rate limited. Lipid-based drug delivery systems (LBDDS) have emerged as a highly suitable formulation strategy to increase the bioavailability of poorly water soluble drugs [5,6]. LBDDS include many diff erent type of drug delivery systems including oil solutions, emulsions, microemulsions, self-emulsifying drug delivery systems (SEDDS) and micellar systems. Such vehicles typically comprise a digestible lipid with (in the case of more complex self-emulsifying formulations) a blend of surfactants, co-surfactants and potentially co-solvents. SEDDS, in particular have garnered signifi cant interest, particularly given the commercial success of the soft gelatin capsule formulation of cyclosporin A, (Mueller et al., 1994). The principal characteristic of a SEDDS is their ability to form fi ne oil-in-water emulsions upon mild agitation following dilution by aqueous phases.SEDDS which form stable nano-emulsions, with droplet sizes less than 300nm, are sometimes referred to as Self Nano- emulsifying drug delivery systems (SNEDDS) [7]
The flagship commercial LBDDS was that of cyclosporine which revolutionized therapy of this potent immuno–suppressant, which had previously displayed low and unpredictable oral absorption, which restricted its widespread use. Since the commercial launch of Neoral® in 1995, it is now estimated that currently up to 4% of all drug products marketed worldwide are formulated as lipid-based formulations [8]. The table below summarized some of the more notable commercial successes with LBDDS and some of the more recent examples of LBDDS in development
Increased Drug Solubilization in Gastrointestinal Fluid
The well-known effect of enhanced bioavailability of many poorly soluble drugs in the fed-state, where the enhanced absorption is ascribed to the co-ingested dietary lipids, is ample evidence of the benefi cial role that lipids can have on drug absorption. One of the reasons for an enhanced food effect relates to stimulation of endogenous biliary amiphiles secretion, such as bile salts and phospholipids, promoting drug solubilizing within the GIT. LBDDS are designed to mimic this effect where the drug is solubilized within the various colloidal lipid structures that can exist in LBDDS and therefore bypasses the drug dissolution step that is required when drug is administered in a crystalline solid- state. In addition, of the lipid excipients, which display surface active properties facilitate interactions and assimilation of drug into the endogenous vesicular and micellar species that form in the GIT. The mechanism of action of LBDDS may also be described using the ‘spring and parachute’ concept, as applied to supersaturating drug delivery systems [16]. LBDDS result in the formation of a supersaturated (i.e. enhanced solubilized) state of the drug on dispersion in gastric fluid, (i.e. ‘spring’) and by facilitating assimilation into post digestive mixed micellar state, the supersaturated state is maintained, within the GIT i.e. ‘parachute’ approach.
Increased Intestinal Membrane Permeability
The inherent permeability enhancing abilities of various bile salts and end-products of lipid digestion (i.e. fatty aids and monoglycerides) in the GIT are well known [17,18]. While it has long been believed that this effect was, at least in part, related to the micellar state increasing passage of the solubilized drug through the aqueous boundary layer, there is now increasing evidence of the bioactive nature of many lipid excipients on intestinal membrane function [19]. Lipid excipients have been shown in in vitro cell lines and in situ intestinal models to have a variety of diverse physiological actions that influence the overall absorption process including; increasing transcellular flux, inhibiting efflux mechanism and altering tight junction integrity [19-21].
Promotion of Lymphatic Uptake
Following absorption of drug into the enterocyte, drug may gain access to the systemic circulation via either the blood capillaries (i.e. portal route) or via lymph capillaries i.e. intestinal lymphatic route. The portal blood represents the major absorption pathway for the vast majority of orally administered drugs as it has a high capacity to transport both water soluble and poorly water soluble compounds. Specifi cally, access to the intestinal lymph (as opposed to the portal blood) is provided for by the more ‘open’ structure of lymph vessels. The majority of drug transported via intestinal lymph is associated with lipoproteins secreted by the enterocyte [22]. Lymphatic transport has been demonstrated for a range of drugs including cyclosporin, mepitiostane, probucol, halofantrine and a range of lipophilic prodrugs [23]. While all the factors influencing lymphatic transport have still to be elucidated it is known that the physiochemical properties of the drug, the interaction of the drug with the physiochemical milieu in the intestinal lumen during lipid absorption, and the chemical nature of any co-administered vehicle are crucial. Hence for drugs which display highly lipophilic characteristics (e.g. log P >5) or signifi cant solubility of intestinal triglyceride (e.g. >50mg/g in triolein), extensive intestinal lymphatic transport has been demonstrated, and in some cases the extent of lymphatic transport is the primary determinant of oral bioavailability for these compounds e.g. Halofantrine [24]. LBDDS, and in particular LBDDS that stimulate production of triglyceride rich lipoprotein in intestinal cells, have been shown to improve the bioavailability of poorly absorbable drugs by increasing by the intestinal lymphatic uptake of the drug [25,26].
Formulation Design Considerations and Predicting In Vivo Performance of LBDDS
Much attention has focused on lipid emulsion pre concentrates (i.e. SEDDS), which can be prepared by physical mixing of lipids excipients, to form stable formulations that are suitable for encapsulation as unit dosage forms (i.e. capsules). SEDDS are usually formulated empirically, though there are some useful guidelines, which have emerged by characterization of the properties of successful formulations [27,28].
Factors Influencing Formulation Design and Excipient Selection
A clear understanding of the characteristics of lipid excipients, and how they influence performance in vivo is desirable to guide formulation design strategies.
Excipient Selection
A key consideration in oral formulation design is the safety and regulatory status of the lipid excipients. In terms of oral formulations, digestible food-grade oils are the preferred oil phase. However, limitations with regard to variability due to their natural origin, low solvent capacity for certain drugs and poor emulsifi cation properties often restrict the use of natural food grade oils. Hydrolysed oils and modified oils, such as polyglycolysed glycerides, have been employed to overcome these issues [29]. With regard to fatty acid type, long chain oils are favored in formulations where targeting uptake via the lymphatic route [22]. However, medium chain oils may be benefi cial for drug solubilization because they are more easily emulsifi ed in aqueous medium [30]. Non-ionic surfactants are considered the surfactants of choice for LBDDS because of their low oral toxicity [31]. The choice of surfactants, or blend of surfactants, will depend on the HLB of each surfactant, which in turn will govern the ability of the surfactants to form a stable nano-emulsion. Of the available surfactants, polysorbates are particularly useful because of their availability in a wide range of HLB values [30] and their good oral acceptability [32]. Other popular surfactants for lipid-based formulations include polyoxyethylene castor oil derivatives and Polyoxylglycerides.
The ability to self-emulsify in a large field of dilution with water
The ability of a SEDDS to be diluted is essential for its use as a drug delivery vehicle since, after administration, it will be diluted by intestinal media. Craig et al., (1996) found that the self-emulsifying behavior was shown to be dependent on the polarity of the oils, with polar hydrophilic oils (i.e. high HLB) displaying the clearest tendency to self emulsify forming o/w emulsions [33]. Similarly, the HLB of the surfactants and the choice of co-solvents can also influence the ability to self emulsify. Evaluating the impact of dilution using biorelevant media (e.g. simulated gastric fluid, FaSSIF) is therefore an important in vitro characterization study.
The droplet polarity, size and size distribution formed on dilution
Larger droplets are less stable than smaller droplets due to their larger area to volume ratio, and so will tend to grow at the expense of the smaller droplets. Droplet size may also affect the rate and extent of drug release (Tarr & Yalkowsky, 1989). The polarity of the oil droplets is governed by the proportion of oil and surfactants, the HLB, the chain length and degree of unsaturation of the fatty acid, the molecular weight of the hydrophilic portion and the concentration of the emulsifier.
Significance of digestibility
In many cases it is favourable to choose an oil phase that undergoes digestion, as the digested products of the oil phases are believed to enhance the solvent capacity of the final mixed micellar phases that forms in vivo [34]. Mixed micelles containing long chain fatty acids have been shown to display a greater solvent capacity than the corresponding simple bile salts [35]. In addition, the surfactants themselves may also undergo digestion, to varying degrees, which will also influence the solvent capacity of the final pre-absorptive mileau. Therefore assessment in an in vitro lipolysis model is considered of paramount importance to assess the likely impact of digestion in vivo. For example formulation of oils with surfactants may affect digestibility overall, as hydrophilic surfactants, such as Cremophor RH40, may inhibit lipolysis, and as a result one of the mechanisms facilitating in vivo drug uptake may be compromised [36]. The in vitro lipid digestion models have been extensively reviewed and they are discussed in detail elsewhere [37,38].
Solvent capacity of the LBDDS to solubilize the dose
A key design characteristic of a LBDDS is that the dose of drug required is adequately solubilized within the amount of lipid that is present in the unit dosage form (i.e. in the capsule) but also that on dilution with intestinal media the drug is adequately solubilized within the emulsion that forms. As previously stated, the use of biorelevant media in evaluation of this effect is more likely to provide a meaningful prediction of the likely impact in vivo.
Loss of solvent capacity on dilution and/or digestion
The fate of the drug following dilution and digestion of the LBDDS, prior to absorption, will greatly influence the in vivo performance. If there is a loss of solvent capacity during intestinal transit resulting in drug precipitating, this precipitate will then need to undergo dissolution if absorption is to take place. Assessing the risk of precipitation, and the potential impact, if any on overall bioavailability, remains a problematic issue and further optimization of in vitro precipitation test methods are required. In general it is believed that the likelihood of drug precipitation upon dispersion/digestion is higher for formulations that contain relatively higher amount of water soluble surfactants (HLB>14) and/or co solvent. i.e. LFCS Class IV systems. This is an important consideration also for the choice of digestible lipid excipients, given that the capacity of the pre-absorptive milieu to maintain the drug in a solubilized state may vary if the digestible surfactant (exogenously derived) are digested prior to the drug being absorbed.
In summary, the formulation design of LBDDS, and in particular ensuring appropriate excipient selection to ensure a successful in vivo performance can be challenging. Choosing formulation excipients can therefore involve a balance to ensuring adequate solvent capacity to solubilize the entire dose on initial dilution in the stomach, while also ensuring that the formulation lipids during processing into the final micellar state (which has undergone further dilution and digestion) will ensure adequate solvent capacity of the drug. In reality, this balance is currently poorly understood and there is a considerable risk of precipitation of drug during the various intermediate stages of drug transfer from the nano-emulsion state to the micellar state. Importantly, the extent of precipitation will be formulation dependent, but further studies are required to fully predict the likelihood of precipitation and/or guide the appropriate formulation excipient selection.
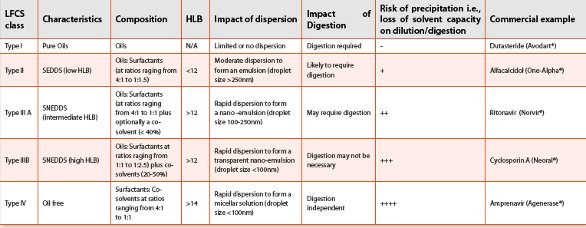
Lipid Formulation Classification Scheme (LFCS)
The Lipid Formulation Classification System (LFCS) has been devised based on similar performance characteristics to those outlined above [28,39]. The LFCS provides a framework to consider key factors (type, digestion, precipitation) that may influence in vivo performance, and while not yet a completely predictive tool, provides a very useful starting point for rational formulation design of LBDDS.
Emerging Applications of Lipid-based Formulations
There have also been some newer formulation approaches designed to further enhance the range LBDDS which are summarized below.
Supersaturable SEDDS
(S-SEDDS) S-SEDDS are SEDDS pre-concentrates that contain a precipitation inhibitor, such as HPMC, that is added to reduce the rate of precipitation from the supersaturated state (i.e. prolong the ‘parachute’ effect) relative to a conventional SEDDS. The S-SEDDS approach has been successfully demonstrated to enhance bioavailability for Paclitaxel, versus a SEDDS control [40].
Nano-structured Liquid Crystalline Phases (Cubosomes)
It has been recently reported that self assembled lipid based liquid crystalline structure, refereed to as Cubosomes, could be effectively utilized as a sustained delivery after oral administration [41]. Cubosomes formed from phytantriol (non digestible lipid) were able to sustain the absorption of cinnarizine over approximately 48 hours, resulting in improved bioavailability, relative to a digestible emulsion formulation.
Scale Up & Large-scale Manufacturing Considerations
While the key advantages of LBDDS have been mentioned in the context of enhancing the oral bioavailability of poorly soluble drug candidates, there are indeed other significant advantages of this formulation approach including the simplicity of the formulation process (i.e. low temperature mixing of lipid excipients), the stability of the system, and ease of scale up. The most common process for preparation of unit dosage forms of LBDDS is using encapsulation technology. Soft gel capsules are single unit solid dosage forms encapsulating a liquid or semisolid fill, which are formed, filled and sealed in a one-step operation using a rotary die process. The technology is available from a number of specialist companies and has been reviewed elsewhere [42,43]. While the technology for filling of hard capsules, which are formed separately and supplied for filling, was extensively used for dry powder/particulate dosage forms, newer advances in both shell type and manufacturing technology has extended its application for liquid fills. Soft gelatine capsules tend to contain higher level of moisture and plasticizers, which is relevant given that some of the lipid excipient can lead to loss of moisture and brittleness of the capsule shell. However newer hard gelatine capsules grades are now available with excellent compatibility for lipid excipients. A key advantage of the hard capsules over soft capsules is the entire process of filling/packaging is shorter.
There have been a number of novel developments in terms of shell material, particular in moving away from gelatine based material to materials which are not derived from animal sources such as plant polysaccharides. In addition softgels may also be coated with a variety of polymers for modified release applications e.g. enteric coated soft gel capsules. Another advantage of the soft gel technology is the ability to produce liquid filled capsules of much smaller sizes e.g. 1-5mm mini-capsules, compared to hard gelatine capsules. This presents opportunities for providing a single capsule containing different populations of liquid filled mini capsules, which would be particularly attractive for combination products e.g. where the APIs are non compatible.
Key Challenges LBDDS - to be or not to be?
LBDDS have generated considerable enthusiasm as well as some important reservations within the pharmaceutical industry. This reticence is largely due to an absence of clear guidelines on formulation design and a paucity of information regarding vehicle effects in vivo [44]. However, in light of the increasing trend towards poorly water soluble drug candidates and the clinical and commercial successes of several lipid-based formulations incorporating problem drugs - namely cyclosporine, ritonavir and saquinavir– there has been a renewed interest in this research field. The choice of a LBDDS to development a new lead compound will be greatly influenced by two key issues: firstly whether the API is the right candidate for LBDDS formulation approach and secondly, can the formulation design be adequately addressed in early stage development to minimize the risk of failure in vivo. Some noteworthy recent initiatives that will further help address these two key challenges are summarized below.
Is the Drug Candidate a Good Choice for a LBDDS Formulation Strategy?
A key consideration in development is to predict as early as possible in the preclinical stage whether a lipid based formulation strategy is a good match for the lead compound. While LBDDS strategies have been successfully applied to a range of poorly soluble drugs, it may be pertinent to look at even more specific physiochemical properties of the drug to further increase the likelihood of a successful outcome. As a general approach, assessing the solubility of the lead candidate in lipids will provide very useful information. Additionally, although the form and content of dietary lipids is markedly diff erent to what would be included in a pharmaceutical formulation, pronounced food eff ect bioavailability data can also be viewed as off ering a clear indicator of where a LBDDS may be successfully applied.
Similarly, assessing solubility of the candidate in biorelevant media, will more accurately predict solubilization capacity in vivo and can also be used to predict food effect in vivo [45,46]. In summary therefore earlier stage screening of enhanced solubilization in lipid vehicles/biorelevant media and/or a pronounced food effect data will assist in predicting whether a LBDDS strategy can be successfully applied. Another more recent initiative that will also aid the understanding of which type of formulation strategy may be employed for enhancing oral bioavailability is the recently proposed Developability Classification Scheme [47]. The DCS, which is an extension of the BCS scheme, further classifies drugs in BCS Class II compounds as either Class IIa (Dissolution limited) or Class IIb (Solubility limited). For Class IIA compounds (e.g. Mefanamic acid) even though the drug solubility will be approaching maximal saturation in vivo, formulation strategies utilizing standard conventional solid dosage forms containing crystalline drug may be suitably applied without the need for specialized solubilization technologies. In other words the formulation strategies can be focused on enhancing dissolution rate (e.g. using particle size reduction technologies) rather than enhancing solubility in the intestine. In contrast, for Class IIb compounds (e.g. Griseofulvin), solubility in the GIT is limiting and therefore technologies that enhance solubilization in intestinal media, such as LBDDS, are ideally applied in this scenario.
Can the In Vivo Performance be Adequately Predicted with In Vitro Methods?
A critical aspect of the development of a safe and effective oral medicine is establishment of meaningful in vitro in vivo correlations (IVIVC) i.e. a predictive mathematical model describing the relationship between the in-vitro characteristics of an oral dosage form and the relevant in vivo response. For conventional solid oral dosage forms, it is well established that this in vitro property is generally derived on the basis of dissolution kinetics. However for LBDDS, given that the drug is pre-solubilized, drug dissolution may not be the appropriate predictive marker for in vivo performance. As described previously, a key performance characteristic of a LBDDS relates to whether the drug remains solubilized in the intestine, starting with the emulsified phase that forms initially on dispersion in intestinal fl uids to the final pre-absorptive micellar phases which is formed post digestion. Further development of in vitro models to simulate the process of dispersion, and digestion of the LBDDS within the GIT are required to gain a better understanding of the in vitro and in vivo drug performance. The establishment of a reliable IVIVC models for LBDDS can then serve as a basis of regulatory guidance for the pharmaceutical industry which will further encourage the development of LBDDS for lead compounds. The significance of established in vitro characteristics is also broader than that identifying which formulation excipients to choose, as these characteristics also defi ne the Critical Quality attributes (CQA’s) which determine in vivo performance. This in turn will have important implications in terms of established CQA’s that are part of the overall new paradigm for systematic and risk based approaches in Pharmaceutical development (i.e. ICH Q8), and the emerging ‘Quality-by-Design ‘QbD’ initiatives.
Table 2. The Lipid Formulation Classifi cation Scheme (LFCS) showing typical performance characteristics for LBDDS (adapted from 28,39)
Conclusions
LBDDS provide for precise and convenient unit dosage forms of lipid formulations, and when appropriately designed allow for rapid assimilation with the relevant bile salt structures on dilution/digestion in the intestine, thereby overcoming dissolution rate limitations for lipophilic drugs. There is a current resurgence of interest in LBDDS due to the potential commercial and biopharmaceutical benefits, and a continuing industry trend towards discovery/development of increasingly poorly soluble (and potent) new chemical entities. The emergence of a number of commercial examples, most notably cyclosporin, ritonavir, saquinavir and amprenavir has furthered interest in SEDDS formulations (Table 2). Clearly there are several areas associated with LBDDS in which further knowledge would be desirable and paramount among these topics is the complex interplay between dilution, digestion and risk of precipitation. In order to achieve more widespread use of LBDDS it is necessary to screen drug candidates, particularly BCS Class IIb, at an earlier stage in drug development to assess whether LBDDS represent a suitable formulation strategy. In addition, further studies on a greater number of poorly water soluble drugs are required so as to gain a more comprehensive understanding, both in vitro and in vivo, of the factors influencing in vivo performance
References
- Hauss, D. J. Drugs and the Pharmaceutical Sciences 2007, 170, vii.
- Amidon, G. L.; Lennernäs, H.; Shah, V. P.; Crison, J. R. Pharmaceutical Research 1995, 12, 413.
- Ku, M. S. Aaps J 2008, 10, 208.
- Horter, D.; Dressman, J. B. Advanced Drug Delivery Reviews 1997, 25, 3.
- Humberstone, A. J.; Charman, W. N. Advanced Drug Delivery Reviews 1997, 25, 103.
- Gursoy, R. N.; Benita, S. Biomed Pharmacother 2004, 58, 173.
- Anton, N.; Vandamme, T. F. Pharmaceutical Research 2011, 28, 978.
- Strickley, R. G. in ‘Oral Lipid based formulations’, Hauss, DJ (eds), Drugs and the Pharmaceutical Sciences, Informa healthcare 2007, 1.
- Mueller, E. A.; Kovarik, J. M.; van Bree, J. B.; Tetzloff, W.; Grevel, J.; Kutz, K. Pharm Res 1994, 11, 301.
- Perry, C. M.; Noble, S. Drugs 1998, 55, 461.
- Strickley, R. G. Pharmaceutical Research 2004, 21, 201.
- Araya, H.; Tomita, M.; Hayashi, M. Drug Metab Pharmacok 2006, 21, 45.
- Perlman, M. E.; Murdande, S. B.; Gurnkowski, M. J.; Shah, T. S.; Rodricks, C. M.; Thornton-Manning, J.; Freel, D.; Erhart, L. C. Int J Pharm 2008, 351, 15.
- Shi, Y.; Gao, P.; Gong, Y. C.; Ping, H. L. Mol Pharmaceut 2010, 7, 1458.
- Chiang, P. C.; Thompson, D. C.; Ghosh, S.; Heitmeier, M. R. J Pharm Sci-Us 2011, 100, 4722.
- Brouwers, J.; Brewster, M. E.; Augustijns, P. J Pharm Sci-Us 2009, 98, 2549.
- Swenson, E. S.; Milsen, W. B.; Curtalo, W. J. Pharm Res 1994, 11, 1132.
- Aungst, B. J. J Pharm Sci 1993, 82, 979.
- Griffin, B. T.; O’Driscoll, C. M. Therapeutic Delivery 2011, 2, 1633.
- Cornaire, G.; Woodley, J.; Hermann, P.; Cloarec, A.; Arellano, C.; Houin, G. Int J Pharm 2004, 278, 119.
- Meaney, C. M.; O’ Driscoll, C. M. Int J Pharm 2000, 207, 21.
- O’Driscoll, C. M. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences 2002, 15, 405.
- Porter, C. J. H.; Charman, W. N. Advanced Drug Delivery Reviews 1997, 25, 71.
- Khoo, S. M.; Edwards, G. A.; Porter, C. J. H.; Charman, W. N. J Pharm Sci-Us 2001, 90, 1599.
- Griffin, B. T.; O’Driscoll, C. M. J Pharm Pharmacol 2006, 58, 917.
- Hauss, D. J.; Fogal, S. E.; Ficorilli, J. V.; Price, C. A.; Roy, T.; Jayara, A. A.; Keirns, J. J. J Pharm Sci-Us 1998, 87, 164.
- Constantinides, P. P. Pharmaceutical Research 1995, 12, 1561.
- Pouton, C. W. Eur J Pharm Sci 2006, 29, 278.
- Shah, N. H.; Carvajal, M. T.; Patel, C. I.; Infeld, M. H.; Malick, A. W. Int J Pharm 1994, 106, 15.
- Gershanik, T.; Benita, S. Eur J Pharm Biopharm 2000, 50, 179.
- Swenson, E. S.; Curtalo, W. J. Advanced Drug Delivery Reviews 1992, 8, 39.
- Benet, L. Z. Mol Pharmaceut 2009, 6, 1631.
- Craig, D. Q. M.; Agnes Chan, K. Y.; Khan, N. Pharmacy and Pharmacology Communications 1995, 1, 559.
- Porter, C. J. H.; Pouton, C. W.; Cuine, J. F.; Charman, W. N. Advanced Drug Delivery Reviews 2008, 60, 673.
- O’ Driscoll, C. M.; Griffin, B. T. Adv Drug Deliv Rev 2008, 60, 617.
- MacGregor, K. J.; Embleton, J. K.; Lacy, J. E.; Perry, E. A.; Solomon, L. J.; Seager, H.; Pouton, C. W. Advanced Drug Delivery Reviews 1997, 25, 33.
- Larsen, A. T.; Sassene, P.; Mullertz, A. Int J Pharm 2011, 417, 245.
- Zangenberg, N. H.; Mullertz, A.; Kristensen, H. G.; Hovgaard, L. European Journal of Pharmaceutical Sciences 2001, 14, 237.
- Pouton, C. W. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences 2000, 11 Suppl 2, S93.
- Gao, P.; Rush, B. D.; Pfund, W. P.; Huang, T.; Bauer, J. M.; Morozowich, W.; Kuo, M. S.; Hageman, M. J. J Pharm Sci-Us 2003, 92, 2386.
- Nguyen, T.-H.; Hanley, T.; Porter, C. J. H.; Boyd, B. J. J Control Release 2011, In Press, Uncorrected Proof.
- Gullapalli, R. P. J Pharm Sci-Us 2010, 99, 4107.
- Cole, E. T.; Cadé, D.; Benameur, H. Advanced Drug Delivery Reviews 2008, 60, 747.
- Charman, W. N. J Pharm Sci-Us 2000, 89, 967.
- Kalantzi, L.; Goumas, K.; Kalioras, V.; Abrahamsson, B.; Dressman, J. B.; Reppas, C. Pharmaceutical Research 2006, 23, 165.
- Shono, Y.; Jantratid, E.; Janssen, N.; Kesisoglou, F.; Mao, Y.; Vertzoni, M.; Reppas, C.; Dressman, J. B. Eur J Pharm Biopharm 2009, 73, 107.
- Butler, J. M.; Dressman, J. B. J Pharm Sci-Us 2010, 99, 4940.
Author Biography
Brendan Griffin, Ph.D. is currently a Lecturer in Pharmaceutics at the School of Pharmacy at University College Cork. Having completed his BSc. Pharm. Degree at the School of Pharmacy in Trinity College Dublin (TCD), he proceeded to a Ph.D. in the Pharmaceutics at TCD. After working in the pharmaceutical industry for a number of years (at Elan and Servier), he returned to academia in 2005. He is also Course Director of the MSc in Pharmaceutical Technology and Quality Systems, a masters course designed to train graduates to become a Qualified Person (QP) in the pharmaceutical industry.