Advanced therapy medicinal products (ATMPs) are medicines for human use that are based on genes, tissues or cells. They offer groundbreaking new opportunities for the treatment of diseases and injuries.
ATMPs include a wide variety of product platforms and involve many new technologies in their development, production, and control. Some of the existing regulatory guidelines pose challenges for advanced therapeutics, and need to be flexible to enable the proper development of innovative therapeutics. The flexibility should not be mistaken for leniency or the lowering of product quality standards, but rather in the adoption of meaningful and appropriate approaches and controls. e.g.; some conceptually different approaches are needed for demonstrating comparability of individualized products when manufacturing processes are updated. Individualized therapeutics are made-to-order for each patient and include multiple product platforms, such as individualized neoantigen-specific therapies.
The European Commission issued: EudraLex - Volume 4 – Good Manufacturing Practice (GMP) guidelines, Part IV - GMP requirements for Advanced Therapy Medicinal Products (for adoption by European Commission on 22 November 2017).3 This 88-page guidance is specific to ATMPs. It is a straight forward simple document for use by the industry to delineate what is expected by the manufacturer from the regulators.
The European Commission also issued; EudraLex - Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 2, Manufacture of Biological active substances and Medicinal Products for Human Use (into operation since 26 June 2018), excludes ATMPs. It makes it clear Part IV is the guidance to go to for implementation of ATMPs.2
The United States Food and Drug Administration on January 2020 issued seven guidelines on the subject of “Cellular & Gene Therapy Guidances.”3-9 these guidances do cover the regulatory requirements for this sector of medicinal products, as indicated in each guidance.
Then, when we try to search for ATMPs guidance harmonization and or related terms (ICH - QSEM); the response: “Your search yielded no results!”12 not there yet! ICH did not anticipate the need for harmonization yet?
All the regulatory guidance aside, e.g.; on the industrialization spectrum of gene therapy with some specificity to viral vectors is very broad including both delivery vehicles developed for transient short-term and permanent long-term expression. These types of vectors are represented by both RNA and DNA viruses with either single-stranded (ss) or double-stranded (ds) genomes. The approval, and or under regulatory review of viral vector-based drugs are making progress in the cure and or treatment of certain ailments.
The field of gene therapy has seen some significant progress with nearly 3000 clinical trials conducted by 2017. Not surprisingly, 64.6% of the trials relate to cancer therapy. Furthermore, 10.5% focus on monogenic diseases, 7.4% on infectious diseases, and 7.4% on cardiovascular diseases. Interestingly, nearly 70% of the trials have utilized viral vectors.1 Although, recent developments in gene manipulation methods, such as CRISPR, and more efficient delivery methods for nonviral vectors, viral vectors still remain attractive.
Based on ‘Regenerative Medicine & Rare Disease 2019 report,’ there were 647 clinical trials in rare disease underway worldwide by the end of 2019, i.e.: Ph 1: 252, Ph 2: 353, and Ph 3:42.11
It is clear that with the growth of this medicinal manufacturing segment of the industry with numerous products in development and or under review for approval by respective regulatory bodies, and an expanding number of organizations involved in ATMPs (Cell and Gene Medicinal Products and Tissue Engineering) manufacture, the demand for streamlined manufacturing, a better understanding of regulatory requirements, knowledgeable operational and regulatory staff and furthermore patient needs are key goals and an important vision for any drug product manufacturing organization to be striving for to deliver the most effective drugs to the patients.
By establishing more consistent practices across manufacturing, logistics, compliance, and many other areas, the industry will be able to overcome some of the current growing pains it is experiencing.
Standardized and robust manufacturing platforms will need to be established, and a global network of facilities will be required to serve the patient globally at a local level at their origin. In addition, fully optimized and robust processes are needed, processes that can be mass customized.
Cell and gene therapy innovators, manufacturers, providers and their contract partners need to focus on manufacturability and process industrialization in the development cycle and avoid technical development bottlenecks. Developers need to be concerned with lean and agile manufacturing that can greatly benefit from working with regulatory, and operational excellence programs to bring about the most efficient and compliant manufacturing and process application specifically developed to deliver this type of product to the patient.
Support for the development and manufacture of safe and effective regenerative medicines and advanced therapies worldwide is required. The combination of therapeutic developers, major medical centers and research institutions, patients’ organizations, tool and technology providers, and regulatory enforcement bodies are key to the successful delivering, streamlining, and integration of processes to provide patients with effective and safe drug products delivered by this segment of the industry.
As stated in: EudraLex - The Rules Governing Medicinal Products in the European Union, Volume IV, and Annex II:
“Reasons for changes: Annex 2 of the GMP Guide has been revised as a consequence of the restructuring of the GMP Guide, new manufacturing technology and concepts, the increased breadth of biological medicinal products to include several new product types such as transgenic derived products and the Advanced Therapy Medicinal Products, (ATMPs) together with associated new legislation. The GMP guidance drawn up for the latter products is to meet the requirements of Article 5 of Regulation (EC) 1394/2007 of the European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/20041 and to align with Part IV of Annex I to Directive 2001/83/EC, as introduced with Commission Directive 2009/120/EC of 14 September 2009 amending Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use as regards to advanced therapy medicinal products.”3
Manufacturing Flexibility
Capacity planning for ATMPs and traditional biologic therapies share the same challenging landscape of unknown regulatory timing, uncertain demand, and highly variable reimbursement decisions. A good well-thought out detailed master plan reduces the risk as products progress from Phase I to Phase III clinical and ultimately commercial launch (or patient use). As with traditional biologics (allogeneic), changes in product demand, process improvements, or increases in automation implementation, and continued process improvements during the clinical phases will challenge the attributes around manufacturing flexibility as well as its surroundings (e.g.: facility, equipment, etc.).
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
ATMPs could be manufactured in a multi-product-based environment that is designed to address the potential risk factors normally associated with this type of manufacturing. Risk factors include product contamination and cross contamination, product mix-ups, human errors, and documentation errors.
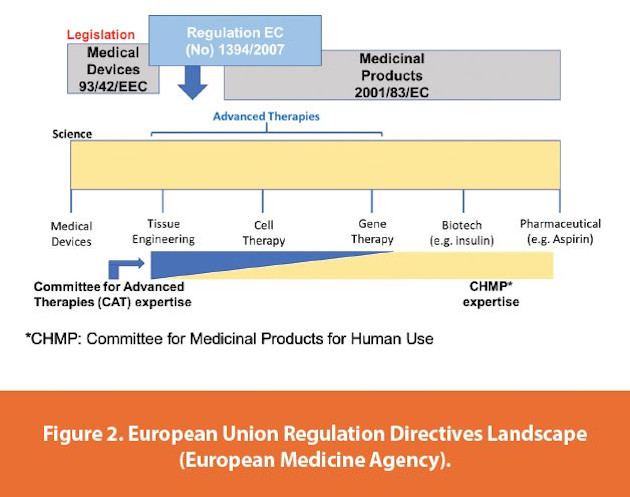
Current regulatory focus around flexibility remains on product protection. As an example, the European Union (EU) clearly defines expectations around concurrent manufacturing and batch/product protection in its guidance.3 The guidance and related directives classify biological medicinal products as delineated in Figure 1 and the applicable directives as delineated in Figure 1 and 2.
Regulatory Considerations
As defined by Regulatory guidances; ATMPs can be classified into three main types (Figure 1 and 3):
- Gene Therapy Medicines: these contain genes that lead to a therapeutic, prophylactic or diagnostic effect. They work by inserting ‘recombinant’ genes into the body, usually to treat a variety of diseases, including genetic disorders, cancer or long-term diseases. A recombinant gene is a stretch of DNA that is created in the laboratory, bringing together DNA from different sources.
- Somatic-Cell Therapy Medicines: these contain cells or tissues that have been manipulated to change their biological characteristics or cells or tissues not intended to be used for the same essential functions in the body. They can be used to cure, diagnose or prevent diseases.
- Tissue-Engineered Medicines: these contain cells or tissues that have been modified so they can be used to repair, regenerate or replace human tissue.
In addition, some ATMPs may contain one or more medical devices as an integral part of the medicine, which are referred to as combined ATMPs. An example of this is cells embedded in a biodegradable matrix or scaffold.3
Since 2017, FDA has approved few regenerative medicine (cellular and gene therapy) products, reflecting continued advancements and a focus on the ATMP field. On July 11, 2018, the agency issued new guidance (draft) documents on disease-specific gene therapy products, which included a guidance on gene therapy products for hemophilia treatment and guidances for retina disorder and rare disease gene therapies that was issued final January 2020.8-10 In addition, with global input from stakeholders, FDA has updated three existing guidance documents that address manufacturing issues related to gene therapy: Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs),5 Testing of Retroviral Vector-Based Gene Therapy Products for Replication Competent Retrovirus (RCR) During Product Manufacture and Patient Follow-up,7 and Long Term Follow-Up (LTFU) After Administration of Human Gene Therapy Products.6 The review of gene therapy products was based on the RCR and LTFU guidance documents previously issued by FDA in November 2006 and the CMC guidance document issued in April 2008.
Figure 4, describes the European Union regulatory pathways for ATMPs, the usual sequence in which procedures are requested by applicants. Note that all procedures can be requested at any time during development of advanced therapy medicinal product.
CMC and Manufacturing of Viral Vectors
To ensure that analysis, production, and quality of viral vector category products is robust and cost-effective, there are a few factors that need to be taken into consideration and explored.
Viral vector characterization and manufacturing presents a core challenge in the commercialization of ATMPs with pressure mounting on CMC, analytics and manufacturing to keep up with accelerated development times and cost effectiveness during the lifecycle of clinical trials and manufacturing process.
More than two decades of experience has generated a substantial amount of data on the safety of retroviral vectors in clinical applications for gene therapy, including experience with different vector designs, vector producing cells, replication competent retrovirus (RCR) detection assays, and lack of positive results from RCR testing of vector lots, ex vivo transduced cells, and patient samples collected during monitoring. These data have provided the basis for public discussions, including Retroviral Breakout Sessions at the 1996 and 1997 FDA/National Institutes of Health (NIH) Gene Therapy Conferences, the 2010 Cellular, Tissue, and Gene Therapies Advisory Committee meeting, and the 2014 American Society of Gene and Cellular Therapy (ASGCT) Breakout Session on Replication Competent Virus. In addition, FDA scientists published an evaluation of RCR testing methods associated with the use of retroviral vectors. During this time, the gene therapy community has improved retroviral vector design to reduce the likelihood of generating RCR during the manufacturing process.7

Development and manufacturing of ATMPs face challenges during production, characterization and quality control of vector-based gene therapies, on rapid CMC development, product and process characterization, upstream and downstream bioprocessing and considerations for large-scale manufacturing.
In development of viral vectors multiple variables must be determined to forecast the demand and capacity for ATMPs. These variables include the projection of the number of clinical trials, patient enrollment in those trials, and viral vector requirements to fulfill the trials. A forecast of the demand requires information about AAV and lentiviral vector manufacturing yields and platforms in use to determine manufacturing capacity required to fulfill vector demands figures.20
Personalized Oncology Vaccines
From development to commercialization, genomic sequencing and bioinformatics understanding are key to development of mRNAbased personalized oncology vaccines (POVs). These custom-tailored vaccines are designed based on each patient’s particular tumor mutations (neoantigens), with the goal of inducing high-affinity immune T-cell responses against cancer.
DNA is extracted from an individual patient’s tumor cells and sequenced. By comparing the sequences of the patient’s tumor mutations with germline DNA from normal cells, tumor mutations are identified.
Then, using proprietary algorithms that evaluate the immunogenic potential of the tumor mutations in the context of the patient’s human leukocyte antigen (HLA) type, the neoantigens most likely to elicit an immune response are selected and incorporated into a vaccine.
This is truly personalized medicine akin to cell and gene therapy and presents unique challenges and opportunities in the CMC cGMP environment. How the one patient one batch concepts are adapted in the clinical stage and what that looks like advancing into the commercial stage is the next challenge to overcome.
Cell Therapy
Over the last decade, the field of cell therapy has expanded rapidly. However, while there are now more cell therapies on offer than ever before, there are a number of factors that are still preventing them from becoming widely used.
Cell therapy holds enormous promise for treating many different diseases. It has also been the cause of controversy regarding the source of the cells.
Some forms of cell therapy, such as hematopoietic stem cell transplantation to treat certain types of cancer, have been around since the 1950’s. While the potential of other types of cell therapy has been known for a while, it has only been in recent years that the field has really started to expand.
The manufacturing and the cost of manufacturing has a large impact here. If you could streamline that whole manufacturing process, improve logistics, have a better controlled supply chain, increase the yield, that will obviously impact significantly in terms of the cost of cell therapy (End-to-End) and reduce the complexity of getting the product to the patient that will help bring down the cost.
Conclusion
Are we there yet? As you scan the spectrum and the breadth of this sector (ATMPs), you realize the field is vast, multi-faceted, complicated and there is not much similarity to harmonize clinical trials and manufacturing. There are a multiplicity of technologies and methods to each medicinal therapy in the segment, and they are moving forward independently. Progress has been made, fast in some instances and slow in others. Capitalizing on experience gained and data obtained from one type of therapy to the other is not conducive due to the nonlinearity between core challenges in the commercialization of ATMPs with pressure mounting on CMC, analytics and manufacturing to keep up with accelerated development times and cost effectiveness during the lifecycle of clinical trials and manufacturing processes. As seen from the EU-EMA regulation and guidances3 and US-FDA regulation and guidances, we see approaches taken are different. The FDA guidance is specific to a segment of the manufacture of a medicinal therapy product, but, the EU-EMA, takes a generalized approach avoiding specific description of the drug substance-drug product specificity in the guidance. We are making progress and some drug products are getting to the patient but we still have a long way home.
References
- Beitelshees M., Hill A., Rostami P., Jones C.H., Pfefifer P.A. Pressing diseases that represent promising targets for gene therapy. Discov. Med. 2017; 24:313–322. [PubMed] [Google Scholar]
- Volume 4 EU guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use; Annex 2 - Manufacture of Biological active substances and Medicinal Products for Human Use, 26 June 2018.
- EudraLex - Volume 4 - Good Manufacturing Practice (GMP) guidelines, Part IV – GMP requirements for Advanced Therapy Medicinal Products (for adoption by European Commission on 22 November 2017.
- Interpreting Sameness of Gene Therapy Products Under the Orphan Drug Regulations; Draft Guidance for Industry 1/2020
- Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs); Guidance for Industry 1/2020
- Long Term Follow-up After Administration of Human Gene Therapy Products; Guidance for Industry 1/2020
- Testing of Retroviral Vector-Based Human Gene Therapy Products for Replication Competent Retrovirus During Product Manufacture and Patient Follow-up; Guidance for Industry 1/2020
- Human Gene Therapy for Hemophilia; Guidance for Industry 1/2020
- Human Gene Therapy for Rare Diseases; Guidance for Industry 1/2020
- Human Gene Therapy for Retinal Disorders; Guidance for Industry 1/2020
- Regerative Medicine & Rare Disease 2019 report (Alliance for Regenerative Medicine, ARM)
- https://www.ich.org/ search engine
- Robert Dream, Jeffery Odum; Impact of ATMP Manufacturing on Process Equipment and Facility Design; BioPharm International, Volume 31, Issue 11, pg. 30–34.
- Article 17 of Regulation (EC) 1394/2007, https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:324:0121:0137:en:PDF
- Article 56 and 57 Regulation (EC) 726/2004, https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2004_726/reg_2004_726_en.pdf
- The Innovation Task Force (ITF), https://www.ema.europa.eu/en/documents/presentation/presentation-innovation-task-force-itf-dr-marisa-papaluca-amati_en.pdf
- Procedural advice on the certification of quality and nonclinical data for small and medium sized enterprises developing advanced therapy medicinal products, https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/procedural-advicecertification-quality-non-clinical-data-small-medium-sized-enterprises-developing_en.pdf
- How scientific advice works, https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-advice-protocol-assistance/how-scientific-advice-works
- Approved Cellular and Gene Therapy Products by US-FDA; https://www.fda.gov/vaccinesblood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapyproducts. “Below is a list of licensed products from the Office of Tissues and Advanced Therapies (OTAT); Approved Products
- ALLOCORD (HPC, Cord Blood), SSM Cardinal Glennon Children’s Medical Center
- CLEVECORD (HPC Cord Blood), Cleveland Cord Blood Center
- Ducord, HPC Cord Blood, Duke University School of Medicine
- GINTUIT (Allogeneic Cultured Keratinocytes and Fibroblasts in Bovine Collagen), Organogenesis Incorporated
- HEMACORD (HPC, cord blood), New York Blood Center
- HPC, Cord Blood, Clinimmune Labs, University of Colorado Cord Blood Bank
- HPC, Cord Blood - MD Anderson Cord Blood Bank, MD Anderson Cord Blood Bank
- HPC, Cord Blood - LifeSouth, LifeSouth Community Blood Centers, Inc.
- HPC, Cord Blood - Bloodworks, Bloodworks
- MLYGIC (talimogene laherparepvec), BioVex, Inc., a subsidiary of Amgen Inc.
- KYMRIAH (tisagenlecleucel), Novartis Pharmaceuticals Corporation
- LAVIV (Azficel-T), Fibrocall Technologies
- LUXTURNA, Spark Therapeutics, Inc.
- MACI (Autologous Cultured Chondrocytes on a Porcine Collagen Membrane), Vericel Corp.
- PROVENGE (sipuleucel-T), Dendreon Corp.
- YESCARTA (axicabtagene ciloleucel), Kite Pharma, Incorporated
- ZOLGENSMA (onasemnogene abeparvovec-xioi), AveXis, Inc.”
- Joseph Rininger, Ashley Fennell, Lauren Schoukroun-Barnes, Christopher Peterson, and Joshua Speidel; “Capacity Analysis for Viral Vector Manufacturing: Is There Enough?,” BioProcess International, December 17, 2019.
- Regulatory Considerations for Human Cells, Tissues, and Cellular and Tissue-Based Products: Minimal Manipulation and Homologous Use. Guidance for Industry and Food and Drug Administration Staff. November 2017, Corrected December 2017. https://www.fda.gov/media/124138/download