Introduction
A lack of data integrity often is “just fraud,” says Howard Sklamberg, FDA deputy commissioner for global regulatory operations and policy. FDA relies on company information documenting adherence to cGMPs, he explained at a July 2014 conference sponsored by the Food and Drug Law Institute. Yet almost all recent warning letters cite evidence of altered and falsified records. If data are “knowingly incorrect, we take that very seriously,” Sklamberg stated, expressing dismay that some manufacturers still fail to remedy record-keeping problems despite repeated warnings from the agency. For Indian pharmaceutical companies supplying generic drugs to the U. S. market this has meant import alerts excluding their drug products from the U.S. (Wechsler, 2014).
What is a Lack of Data Integrity
Many people in the pharmaceutical industry are confused by the concept of data integrity. Data integrity is comprised of these following broad actions to hide test failures and/or manufacturing deviations:
- Omission of data
- Errors in data recording
- Changing data
- Deleting data
Destroying data These actions may be both unintentional and intentional, representing GMP violations that can have civil and criminal consequences to the company and seriously damage the company’s business.
Examples of a lack of data integrity in chemical testing include:
- Not documenting activities or failing to document activities at the time performed (pre- or post-dating)
- Testing and discarding failing data
- Testing and only reporting passing data.
- Fabricating data
- Using previously generated data
- Not following test procedures and sampling plans
- Missing, altered or raw data not captured on test report or batch records
- Memorized or recorded data on loose pieces of paper
- Electronic records changed without an audit trial
Why Has Data Integrity Become a Hot Topic?
Beginning in 2014 there was a marked increase in warning letters to Indian drug manufacturers addressing data integrity as the FDA uncovered problems during regulatory inspections (Unger, 2017). This increase is illustrated in Table 1.
Table 1. The Number of FDA Warning Letters Issued Addressing Data Integrity
Is the Issue Limited to Indian and Chinese Pharmaceutical Companies?
In 2016, the FDA issued 39 warning letters citing a lack of data integrity (Unger, 2017). The national distribution of warning letters was China 13 (33%), India 9 (23%), U.S.A. 6 (5%), E.U. 6 (15%), Brazil 3(8%) and Japan 2 (5%).
The FDA completed a total of 5,615 GMP inspections of registered drug and device establishments in FY 2015. Of these 4,055 (72%) were domestic inspections and 1560 (28%) were foreign inspections. Based on the recent U.S. GAO-17-143 Report on the FDA’s Foreign Offices and Drug Inspections in FY 2015 there were 842 foreign drug establishment inspections of which 527 (63%) were for GMP surveillance, 250 (30%) for pre-approval and surveillance, 23 (3%) for preapproval only and 42 (5%) for cause. The report highlighted the high frequency that an establishment may have never been inspected. For example, in the FY 2017 catalog were 572 Indian establishments with 33% never inspected and 535 Chinese establishments with 45% never inspected. This suggests that Indian and Chinese drug manufacturing facilities will continue to be cited for data integrity issues into the foreseeable future unless they aggressively address data integrity issues within their companies.
Data Integrity Directed Towards Microbiological Testing
Many of the cited data integrity issues are not associated with microbiology but with chemical analyses, especially High-Performance Liquid Chromatography (HPLC). The norm for microbiological testing is the visual inspection of media for the detection of microbial growth or the enumeration of microbial counts. Typically the results are recorded on paper worksheets or more recently into electronic notebooks. The data, but not always the original microbial cultures, are checked by a second person for completeness and the absence of recording and arithmetical errors. The data are then entered into Laboratory Information Management Systems (LIMS) and approved by a laboratory supervisor. These laboratory procedures place a high reliance on the integrity, experience, and training of the individual analyst, the ability of the peer reviewer or supervisory staff to detect poor data integrity, and the culture of the company to set the highest standard. This type of data could be susceptible to falsification. Microbial methods rarely take advantage of modern analytical technologies.
Contributing factors to reduced data integrity in the microbiology laboratory are the subjectivity of scoring microbial growth in broth, analyst-to-analyst variability in counting colonies on plates, and the inability to perform aseptic manipulations with the contemporaneous recording of data. Often LIMS data fields for many microbial measurements such as < 10 CFU/g, Log 10 counts, and absence of specified microorganisms test results are lacking or inconsistent. For most microbiological tests, the absence of computer data collection, automatic calculations and generation of a finished report lack detail and flexibility.
A broader discussion of the issue of data integrity in the area of microbiology could be useful in terms of setting industry and regulatory expectations. Microbiological data has historically been evaluated and recorded manually by appropriately educated and experienced microbiologists trained in the art of contamination detection and colony counting. Microbiologists must use experience, expertise and judgment for test interpretation, which may lead to subjective and variable interpretation and documentation of test results. While the use of rapid technology, digital image capture and automated plate readers can remove most of the subjective data interpretations and solve data integrity issues, the algorithms used to count colonies or detect contamination are subject to validation limitations. Microorganisms grow at different rates, different conditions, evolve to different colony shapes, sizes, and colors that must be accommodated. The addition of a high quality digital image of a plate is helpful in terms of data integrity (verification of test results, data archival, data retrieval) and verification of the accuracy of results for the manual recording of colonies. Microbiologists may still have to override electronic results if spreading colonies, colonies embedded within other colonies in plate counts, or product precipitation or flocculation in sterility tests makes interpretations by instrumentation inaccurate or more challenging.
Risk Analysis
Many pharmaceutical companies are now evaluating data integrity using standard risk assessment tools such as FMEA (Failure Mode and Effects Analysis). In the opinion of the authors the tests which appear to be the most critical, subjective, and prone to data integrity issues are the sterility test, the gel clot Limulus Ambeocyte Lysate (LAL) bacterial endotoxin assay and any microbial enumeration test which involves counting microbial colonies which covers most of the other compendial tests. The sterility and LAL gel clot assays are subjective in their interpretations. The enumeration test accuracy depends on the interpretation of discreet colony counts by the microbiologist reading the plates.
Using risk assessment tools, the authors believe that data integrity issues can be classified as high, medium and low risk processes.
High Risk
- Analyst records incorrect data
- No second opinion for subjective interpretation of test results
- Analyst performs wrong method or wrong sample preparation
- Missed critical data or incorrect information
- Falsification or lack of authentic data
Medium Risk
- Real time data entry difficulties
- Incomplete data entries
- Chain of custody for samples issues
- Delay in testing, times not documented
Low Risk
- Improper recording of non-critical data
- Lack of real time entry of non-critical data (lot numbers, expiry dates, and recalibration dates.)
GMP Requirements with Respect to Data Integrity
Data integrity is addressed in 21 CFR 211.194 Laboratory Records where the GMP requirements are defined. Recently four guidance documents addressing data integrity have been written by the U.K. Medicines & Healthcare Products Regulatory Agency (MHRA), Parenteral Drug Association (PDA), U.S. Federal Food and Drug Administration (FDA), and Pharmaceutical Inspection Co-operation Scheme (PIC/S). The documents are:
- 2015 MHRA GMP Data Integrity Definitions and Guidance for Industry
- 2016 PDA Elements of a Code of Conduct for Data Integrity in the Pharmaceutical Industry
- 2016 Draft Guidance for Industry – Data Integrity and Compliance with CGMP
- 2016 PIC/S Guidance Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments
Why These Documents Inadequately Deal with Data Integrity Issues in Microbiology
On April 14, 2016 the FDA published a Draft Guidance for Industry on data integrity that emphasized computer data management systems but not specifically microbiological testing. The document defined data integrity as to the completeness, consistency and accuracy of data. They used the acronym ALCOA which stands for Attributable (A), Legible (L), Contemporaneously recorded (C), Original or a true copy (O) and Accurate (A).
On August 10, 2016, a draft PIC/S guidance document was published to provide industry with a “consolidated, illustrative guidance on riskbased control strategies”. The new guidance from PIC/S was applied on a 6-month trial basis by participating regulatory authorities. The authors agree with the practical approach taken by PIC/S.
The PIC/S guidance defines and deals with:
- Data governance systems
- Organizational influences on successful data integrity management
- Specific data integrity principles and enablers
- Data integrity considerations for paper-based and computerized systems
- Integrity considerations for outsourced activities
- Regulatory actions in response to data integrity findings
The guidance wisely states, “Management should aim to create a work environment (i.e., quality culture) that is transparent and open, one in which personnel are encouraged to freely communicate failures and mistakes, including potential data reliability issues, so that corrective and preventative actions can be taken.”
The authors agree that the PIC/S draft guidance, unlike the other regulatory documents, details the various deficiencies linked to data integrity failures that may have varying impacts on product quality (i.e., a risk-based approach to a loss of data integrity):
Critical deficiency (Impact to product with risk to patient health):
- Product failing to meet specification at release or within shelf life. Reporting of a “desired” result rather than an actual out of specification result when reporting of quality control (QC) tests, critical product or process parameters.
Major deficiency (Impact to product with no risk to patient health):
- Data being misreported (e.g. original results “in specification,” but altered to give a more favorable trend). Reporting of a “desired” result rather than an actual out of specification result when reporting of data, which does not relate to QC tests, critical product or process parameters. Failures arising from poorly designed data capture systems (e.g. using Postits® to record information for later transcription) No impact to product; evidence of widespread failure.
- Bad practices and poorly designed systems which may result in opportunities for data integrity issues or loss of traceability across a number of functional areas production, QA, QC, etc., though individually, each violation “has no direct impact to product quality”
Other (minor) deficiency (No impact to product with limited evidence of failure):
- “Bad practice or poorly designed system, which results in opportunities for data integrity issues or loss of traceability in a discrete area. Limited failure in an otherwise acceptable system”
Application of ALCOA Principles to Microbiological Data Integrity Issues
Applying the ALCOA principles to microbiology testing may on the surface seem to be a common-sense approach to cGMP compliance. However, after applying risk-based principles to the discreet tasks related to microbiological testing, documentation can be difficult and cumbersome. Approaches to applying the ALCOA principles can be minimal, moderate or extreme. Extreme approaches may counteract good documentation practices. A risk-based approach to the criticality and functionality of the task and its documentation should be used. Companies also need to decide if “data” includes ancillary items such as media, reagent lot numbers, and expiration dates, instrument numbers and their recalibration dates, and laminar flow hood numbers. Applying extreme approaches to ALCOA principles for data integrity can result in a second person having to shadow the microbiologist and witness every documented step. Microbial tests also involve the use of aseptic techniques where real-time recording of the tasks cannot be performed without jeopardizing the quality of the results. Manual documentation of hundreds of environmental monitoring, water, or enumerations plates are often batch read with “no growth detected” plates being separated from “growth detected” plates. All documentation is batch recorded at the same time. Do the ALCOA principles require each plate read, documentation, and recording of a second person confirmation all have to occur at the same time? Do the principles require that a second person have to re-enumerate quantitative plates to confirm the accuracy of the colony forming units? The authors believe not. For microbiology testing, many tests are performed over days, even weeks. How do we document teamwork? Frequently multiple individuals work on the same test, performing dilutions, culturing organisms, counting plate colonies, and reading or interpreting results at different times over a number of days? How are ALCOA principles applied? These FDA 483 observations give an insight into the regulatory thinking about data integrity as applied to microbiology. Microbiologists can also evaluate how rapid technologies may be helpful in remediating the problems.
Attributable
The first ALCOA principle is ATTRIBABLE. The data is linked to the person performing the action. Only that person may sign for the work completed. An FDA 483 observation was given to a company which read: “While multiple employees participate in a process step that spans more than one shift, only the last person involved is required to sign the batch record for completion of the activity.” Another observation read: “The inspection documented that all of your QC laboratory computerized instruments (redacted) were found to be stand alone, and laboratory personnel demonstrated that they can delete electronic raw data files from the local hard drive. Your firm deleted multiple data files acquired in 2013 allegedly to clear up hard drive space without creating backups. Your QC management confirmed that there is no audit trail or other traceability in the operating system to document the deletion activity. Furthermore, your analysts do not have unique user names and passwords for the computer and laboratory information systems; your QC analysts use a single shared user identifier and password to access and manipulate multiple stand alone systems.”
While these observations may not apply directly to microbiology laboratories, there is a concern around the inability to attribute a task or data set to a person or group of people. Often hundreds, even thousands of microbiology plates are read. There may be multiple individuals all working together to complete the task spanning more than one shift, or even more than one day. How granular and frequent must the tasks be documented?
The requirements for data integrity were not intended to limit laboratory flexibility or agility. Problems can arise when personnel must repeatedly log in and out of computers for each task completed to document individual contributions to the team. Shared electronic notebooks can help, but care must be taken to disallow and/or track overwrites and accidental omissions. In some ways, paper documentation is easier than electronic documentation. However, electronic documents can be templated, time stamped, and changes tracked, which is a huge advantage. The advantage of rapid technology automatic data capture readers is that they can address many of the data integrity concerns. The use of automated plate readers and rapid technology instruments may come with some logistical issues since one must assure that functions such as audit trails are turned on, and permissions for tasks appropriately assigned in a manner that there is no conflict of interest. The ability to reproduce/retain the raw data in a readable form for archival and retrieval must also be achieved.
Legible
The second ALCOA principle is LEGIBLE. Companies have received observations for obvious use of correction fluids, overwrites/ scribbles on handwritten data sheets, and not having appropriate change controlling procedures in place. This includes access to and use of signature stamps for electronic systems in lieu of handwritten signatures. General data integrity principles have always mandated that data must be readable throughout the data retention period and lifecycle, with permanent and identifiable changes using permanent ink, neatly, and following established date and time formats. Electronic data has additional challenges. The rules apply to all raw data, meta data, and finished data reports which are stored and backed up for archival and retrieval. Many computerized systems store the raw data and use internal software to access, process, and report the information. Computer systems and programs are constantly being updated. Therefore, the retired instrument programs may also need to be archived in a manner that raw data can be retrieved and reprocessed. Computer operating systems may not be able to run the specialized software after many years. This conundrum has yet to be resolved.
Contemporaneous
The third ALCOA principle is CONTEMPORANEOUS. For microbiologists, this may pose the most challenging principle. These challenges include:
- Signing/dating records at the time of activity completion
- Back-dating or pre-dating data sheets
- Limiting access to change date/time of electronic data
- Aseptic manipulations under laminar flow hoods or in isolators including the opportunity to document a microbial test
A recent 483 observation was …. “Specifically, an operator performed the in-process tablet (redacted) testing for the (redacted) mg tablet batch #(redacted) without the batch record or a manufacturing form to document the results contemporaneously. …your operator stated that he records the two weights with (redacted) significant figures into the batch record (located in another room) from memory”. Do tasks or data have to be recorded during microbial testing when activity occurs and how do we maintain aseptic technique while still complying with the requirements for data integrity?
Original
The fourth ALCOA principle is ORIGINAL. This can be difficult for microbiologists. Many microbiologists write the plate counts on the plate. Many isolate zero count plates into one location to document all negative or 0 CFUs later. The authors believe these are common practices, which allow the laboratory operations to remain productive and flexible.
Original data is data as the file or format in which is was generated, preserving the integrity (accuracy, completeness, content and meaning) of the record
- Retain all raw data and printouts
- Original data must be recorded directly into GMP records
- Original data must be reviewed
- Electronic raw data files must be retained
A recent 483 observation stated: “Your firm repeatedly delayed, denied, limited an inspection or refused to permit the FDA inspection. An FDA investigator identified the presence of torn raw data records in the waste area and asked one of your firm’s QA Officers to remove these torn raw data records for the investigator’s review. This QA Officer presented the FDA investigator with approximately 20 paper records, none of which included raw data entries identified in the waste area earlier during the inspection. The FDA investigator then revisited the waste area and found that the raw data records had been removed and placed in a different holding bag.” While this observation is an example of a more egregious breach of data integrity, microbiologists struggle with exactly what is original data. If plate counts are written on the plate and not recorded directly onto a report, are the plates raw data?
Accuracy
The fifth and last ALOCA principle is ACCURACY. This article addresses the Who, What, Where, When, Why and How data represents the complete set of factual data and meta data.
- Data must reflect the action/observation accurately
- All data must be controlled and retained for the lifecycle
- Modifications to the data must be explained if not obvious
- Meta data includes units of measure, method details, audit trail, date/time modifications
Recent FDA 483 observations stated: “Processing each result file individually using different processing parameters generates the data for assay. The second person reviewer is not required to review each of the processing parameters used to generate results.” Another observation is “Only one operator was assigned to evaluate sterility test on the last day of the incubation period. To ensure reliability of the tests, your procedure needs to specify steps that enable objective evaluation, such as assigning two operators for evaluation on the last day.” This implies that the FDA expects a second microbiologist check all sterility test media at the end of the test time.
A third recent FDA 483 observation stated: “Not all laboratory data were recorded accordingly. On 5/25/2017 an FDA microbiologist observed the following: a. “The firm’s microbiologist read the settling plate at location Vial Sealing Area Passive on 5/24/2017 and reported “0” CFU. A FDA microbiologist observed 1 CFU for the same plate.”
The observation that the company reported no CFU and the FDA investigator was finding 1 CFU is disconcerting and may be a serious data integrity issue. The delay in examining the plates may have been a factor - the plates were read 5/24/2017 and the inspection conducted between 5/25 and 6/1/2017. Should one automatically assume that there was falsification of the environmental monitoring results? The authors believe that only the CFUs reported within the recommended incubation time are valid. Since specifications are for a defined incubation period, viewing colonies after the end of the incubation may not be valid. It is possible that a slower growing colony was not visible at termination of the test. It is possible that one opens the plates for a better view to read the plates. One should not automatically infer error or fraud for data which is acquired during a set period of time and the analytes are known to be dynamic at the end of a tests’ duration.
Risk Mitigation
How can the risk be mitigated with microbial test methods? What should and should not industry do in response to data integrity issues? The first step is to acknowledge the limitation of the classical microbial test methods, (accuracy and precision), their dependency on the technical capabilities, honesty of the microbiologists conducting the tests, and supervisors reviewing and approving the tests for data integrity (Table 2). Ultimately the maintenance of data integrity is the joint responsibility of upper management, managerial and supervisory staff, and bench-level microbiologists. The authors do not advocate employing a second microbiologist to review all microbial test results, but potential failures of critical tests should be reviewed contemporaneously, if possible, by a second person. Test result reports should be reviewed by a superior for accuracy and completeness. Outof-specification results should be fully investigated. Unlike the opinion of some FDA investigators, the authors believe that negative sterility tests do not require a second person review.
Table 2. Data Integrity Level of Classic Microbial Test Methods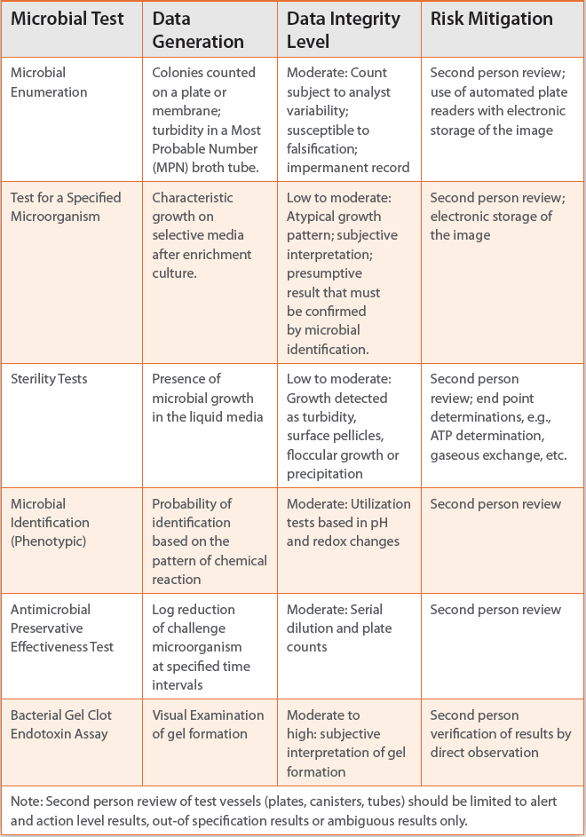
Alternative Microbial Test Methods
Are the risks different with alternative microbial test methods? Will this difference drive the implementation of these newer methods? Alternative microbiological test methods usually depend on analytical signals that are quantitative, more sensitive, and usually more reliable than classical growth-based methods (Table 3). Many alternative methods generate results in real time so can be described as rapid microbial methods (RMM). The signals generating results may not be fully equivalent to colony-forming units counted in a traditional plate count. Analytical methods are typically objective, do not rely the subjective scoring of colonies growing on a plate or turbidity of a broth, and can be captured electronic and archived which means they have inherent data integrity. These characteristics of alternative microbial test methods should help promote their implementation in the pharmaceutical industry. However, many of the data integrity issues associated with chemical assays would likely be applicable to these newer microbial methods.
Table 3. Data Integrity Level of Alternative Microbial Test Methods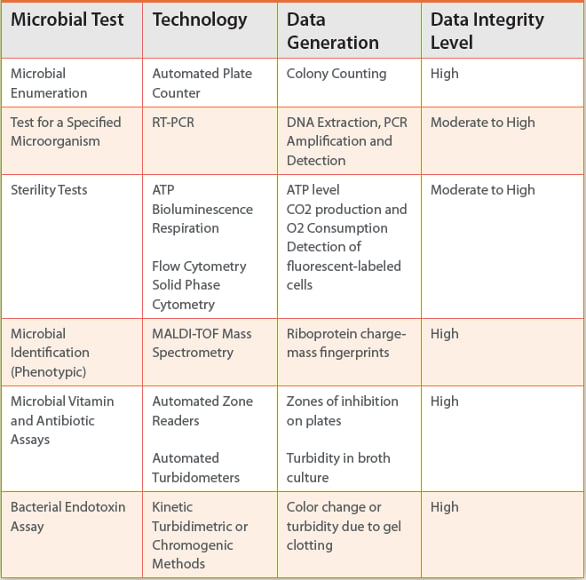
Concluding Statements on the Issue of Data Integrity
The amount of recent regulatory activity in the area of data integrity and the failure of four recently published guidance documents to address microbiological issues is a concern to pharmaceutical microbiologists. The challenge to ensure data integrity in the microbiology laboratory is unique. Computerized record keeping is not as advanced as it is in chemistry laboratories. In most cases contemporaneous data collection is not possible due to the necessity of using aseptic techniques or contamination control. Furthermore, the volume of plates to be read in support of environmental and water monitoring makes second person review of all plates impractical. The authors believe that the right response to this challenge is that second person review be limited to alert and action level results, outof-specification results or ambiguous results only. Completed work sheets should be diligently reviewed by laboratory supervisors and laboratory test failures aggressively investigated. Ultimately, data integrity can only be ensured by the maintenance of high ethical and professional standards throughout a pharmaceutical company.
References
- FDA Draft Guidance for Industry – Data Integrity and Compliance with CGMP www. fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ UCM495891.pdf
- Wechsler, J. Data integrity key to GMP compliance. BioPharm International (September 2014) www.biopharminternational.com/data-integrity-key-gmp-compliance
- Unger, B. 2017 An analysis of FDA warning letters on data governance and data integrity Pharmaceutical Online, Guest Column July 14, 2017 www.pharmaceuticalonline.com/doc/ an-analysis-of-fda-warning-letters-on-data-governance-data-integrity
- U.S. Government Accountability Office GAO-17-143 Report on the FDA’s Foreign Offices and Drug Inspections www.gao.gov/products/GAO-17-143
- PIC/S Guidance Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments 2016 www.picscheme.org/en/publications