Introduction
Monoclonal antibody (mAb) has become one of the fastest growing therapeutic modalities within the pharmaceutical industry, with an increasing number of blockbuster mAb drug products approved over recent years.1 Heterogeneity of therapeutic antibody products as a result of enzymatic/chemical modifications during bioprocessing and storage is well-documented.2 Protein characterization methods using LC-MS, including top-down, middle-down or up, and bottom-up approaches, have been widely used to study antibodies during drug discovery and development.3 In spite of the continuous increase in the resolving power of liquid chromatography (LC) and mass spectrometry (MS), it remains a great analytical challenge to fully characterize the heterogeneity of a therapeutic antibody using a single analytical method. Only middle-up and bottom-up LC-MS methods have shown potential of monitoring multiple quality attributes. Peptide mapping, a powerful tool in characterizing the site-specific post-translational modifications of therapeutic proteins, has been applied in monitoring multiple attributes of antibodies in recent years.4
In comparison to peptide mapping, the application of middle-up LC-MS methods based on limited proteolysis of mAb in the hinge region by papain, lysyl endopeptidase (Lys-C) and pepsin has been relatively limited. This is likely due to the nature of pepsin, papain and Lys-C, which requires a well-controlled condition to yield desirable F(ab’)2 and scFc fragments in a reproducible and complete manner for further characterization and quantitation of product quality attributes.5 IdeS (immunoglobulin-degrading enzyme of Streptococcuspyogenes), a better alternative to the aforementioned proteases, has gained increasing interest in recent years for application in middle-up intact mass for routine antibody characterization as well as product quality monitoring.5-7 IdeS cleaves at heavy chains below the hinge region of all IgG class through a simple but robust digestion procedure, producing F(ab’)2 and scFc fragments, which are typically further reduced using DTT or TCEP to produce three subunits or domains of antibody, e.g. light chain, Fd’ and scFc. Each of the subunits has a molecular weight of ~25 kDa, facilitating the separation of variants of each subunit from its native form, such as oxidation, C-terminal or N-terminal variants.7 An excellent review has summarized the application of IdeS in antibody characterization since it became commercially available.6
In the present study, an LC-MS method was developed to analyze IdeS digests of NIST mAb and a few in-house IgG1 mAbs, with or without further reduction. The capability of this subunit analysis in simultaneous characterization of multiple attributes at domain levels was examined. In addition, the subunit analysis was also examined for potential capability of providing (semi-)quantitative assessment of pH labile modification such as cyclic imide at domain level, or to probe domain-domain interactions that form aggregate.
Experimental Procedures
Four IgG 1 recombinant mAbs from NIST and Biogen were used to develop IdeS-based subunit analysis in this study. All IgG 1 mAbs were expressed in a chinese hamster ovary cell system, and stored at -70 °C in respective formulation buffers prior to analysis.
Sample Preparation for IdeS digest
IdeS digest of IgG1 mAb was prepared by diluting 100 μg of sample to 2 mg/mL with IdeS digestion buffer 50 mM sodium phosphate, 150 mM sodium chloride, pH 6.6. Two microliters of IdeS solution was added and the mixture was subsequently incubated at 37 °C for 30 min. The obtained IdeS digest of IgG 1 mAb can be further reduced by combining 20 μL of the IdeS digest with 40 μL of 8 M Guanidine, 10 μL of IdeS digestion buffer, and 10 μL of 1M dithiothreitol (DTT) in a 0.6 mL centrifuge tube, and incubated at 37˚C for 30-60 min. All samples were prepared fresh on the day of analysis. Upon completion of the sample preparation, the samples were transferred to respective autosampler vials and stored at 2-8˚C for LC-MS analysis.
Liquid Chromatography-Mass-Spectrometry and Data Analysis
The IdeS digest of IgG1 mAb from ~4 μg protein was injected and separated on rapid resolution columns (1.8 μm 2.1 × 100 mm, at 55 °C using an UHPLC system online with a mass spectrometer. The mobile phases consisted of 0.03% TFA in water (A) and 0.024% TFA in acetonitrile (B). The chromatography was carried out using a linear gradient from 10 to 30% mobile phase B in 4 min and from 30 to 40% mobile phase B in additional 20 min at a flow rate of 300 μL/min. A slightly modified gradient was applied to mAb 3 in order to better resolve Fd’ variants from its native counterpart. Data acquisition was controlled by software, with typical instrument parameters for mass spectrometric detection for intact mass analysis of recombinant antibodies applied. Data analysis was performed by protein deconvolution software.
Results and Discussion
The aforementioned IgG 1 antibodies were digested by IdeS, which were then followed by DTT reduction under denaturing condition prior to RP-LC/MS analysis by the UPLC in line with mass spectrometer. The LC-UV profiles and peak ID of the mAbs examined are shown in Figure 1.
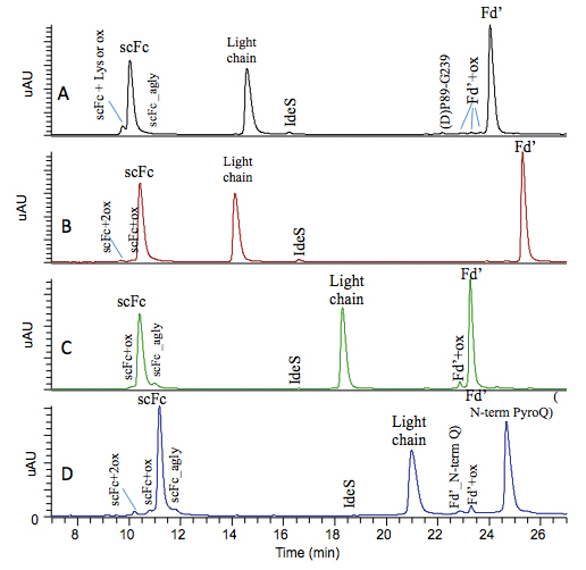 Figure 1. LC-UV profiles of subunit analysis of NIST IgG1 mAb (A) and in-house IgG 1 mAb 1 (B), 2 (C) and 3 (D).
Figure 1. LC-UV profiles of subunit analysis of NIST IgG1 mAb (A) and in-house IgG 1 mAb 1 (B), 2 (C) and 3 (D).The LC-UV profile for NIST mAb subunit analysis obtained is similar to that reported earlier.8 Briefly, oxidized, aglycosylated, and C-terminal lysine variants of scFc are observed at domain level and chromatographically resolved from their native counterparts. Oxidized, and N-terminal variants of Fd’ are also observed at domain level and chromatographically resolved for their native counterparts. D(P) clipped species is observed in NIST mAb in the current study but at a lower level compared to that reported earlier.8 D(P) clipped species can be contributed by prolonged exposure of protein at low pH and high temperature environment,9 such as during the LC separation when protein experiences lower pH mobile phase under elevated column temperature. Therefore, it is expected that different subunit analysis protocol may lead to different level of D(P) clipped species detected. In addition, mass spectrometry provides additional information regarding the N-glycosylation profile in scFc region, and glycation level for each domain. Similar modifications at domain levels were also observed for our in-house IgG 1 antibodies. IdeS-based subunit analysis has demonstrated its capability of simultaneously monitoring multiple attributes of antibodies at domain level, including oxidation, C-/N-terminal variants, aglycosylated scFc, clipped species, Fc-glycosylation, and glycation.
IdeS protease is active in the pH range 5.1–7.6, but with an optimum at pH 6.6,6 making it an attractive tool in monitoring pH labile posttranslational modification such as cyclic imide, or succinimide, an intermediate product of Asp isomerization.10-11 Cyclic imide tends to form at mild acidic pH, but hydrolyzes to form iso-Asp and regeneration of Asp at neutral or alkaline pH, conditions that are commonly used for peptide mapping digestion.12,13 Asp isomerization occurring in protein therapeutics may lead to a decrease in binding activities and thus needs to be closely monitored.14,15 An Asp site on mAb 1 light chain CDR region was confirmed by peptide mapping to be prone to isomerization and cyclic imide modification when stressed at high temperature in acidic pH buffer. IdeS-based subunit analysis was examined as a potential tool in monitoring its cyclic imide level in this study with the aim of providing more accurate assessment of pH labile cyclic imide modification level. IdeS digestion and reduction protocol described above was applied to a mAb 1 sample stressed at 25 °C for 4 months at pH 5.5, along with its control sample, with their LC-UV profile compared in Figures 2Aand 2B. Cyclic imide variant of light chain was not able to resolve from its native domain chromatographically, but can be differentiated by mass spectrometry as shown in their raw mass spectra in zoomed view in Figures 2C and 2D.
 Figure 2. LC-UV profile comparison of IdeS digest of in-house mAb 1 control (A) and stressed sample (B), and zoomed view of light chain mass spectra of in-house mAb 1 control (C) and stressed sample (D).
Figure 2. LC-UV profile comparison of IdeS digest of in-house mAb 1 control (A) and stressed sample (B), and zoomed view of light chain mass spectra of in-house mAb 1 control (C) and stressed sample (D).The cyclic imide modification levels in control and stressed samples assessed by the deconvoluted mass intensity are 3.6% and 20.5%, respectively. In contrast, the cyclic imide modification level in mAb 1 control and stressed samples assessed by lysyl endopeptidase peptide mapping are 0.7%, 10.7%, respectively. Lower cyclic imide modification level detected by peptide mapping than IdeS subunit analysis is likely due to conversion of cyclic imide modification to iso- Asp at slightly alkaline pH used in peptide mapping digestion.12,13 A linear relationship of cyclic imide level based on deconvoluted mass spec intensity was established by mixing mAb 1 control and stressed samples at different ratio and subjecting to subunit analysis with R2 of 0.996 (data is not shown). It is not the intention of the current study to assess the half-life of the pH labile cyclic imide formation under current digestion condition. The IdeS digestion condition was shown to be able to maintain cyclic imide modification to a large degree if not all, evidenced by good accuracy obtained across the linear range (data is not shown). This result implies that IdeS-based subunit analysis can be used to monitor or even quantify domain-specific cyclic imide modification instead of hydrophobic interaction chromatography that was traditionally used.13,15,16
IdeS-based subunit analysis without further reduction may maintain domain-specific structural and conformational information to some degree, and thus was examined as a potential tool in probing the nature of aggregation formation in this study. Aggregate fractions of two mAb 3 samples stressed at different conditions were collected by preparative scale SEC and subjected to IdeS digestion along with control without further reduction prior to reverse phase LC-MS analysis. LC-UV/TIC chromatograms were collected and with only LC-TIC profiles compared in Figure 3, as LC-TIC profile demonstrated more distinct peaks and differences in profiles likely due to different ionization efficiency of mAb domain with different conformation.
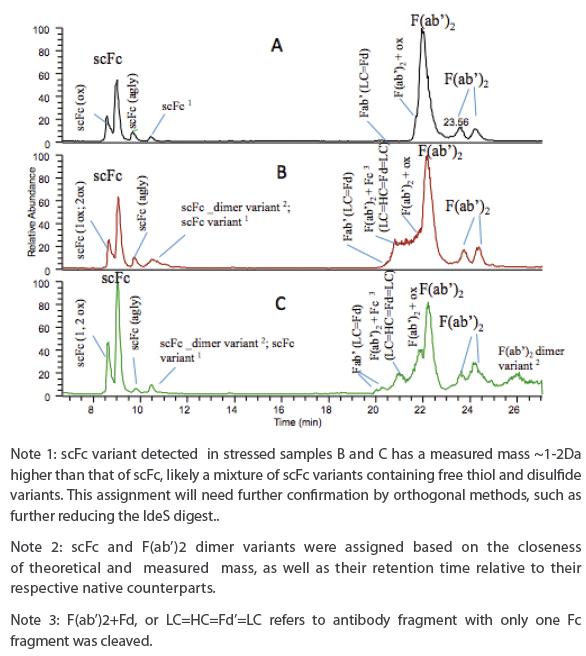 Figure 3. LC-TIC profiles of subunit analysis of aggregate fractions of two stressed mAb 3 samples (B, C) in comparison to control (A).
Figure 3. LC-TIC profiles of subunit analysis of aggregate fractions of two stressed mAb 3 samples (B, C) in comparison to control (A).Several notable differences in LC-TIC profiles are observed between the samples analyzed. Oxidized scFc variants are detected in all samples, but not as well resolved from native scFc as those reduced counterparts as seen in Figure 1D, likely due to the fact that samples was not reduced/denatured and conformation played a role in chromatographic separation. A new peak is shown in all samples eluted after the aglycosylated scFc peak, and a mass species with a deconvoluted mass about 1-2 Da higher than scFc is detected in all samples, likely a mixture of scFc variants containing free thiol or different disulfide linkage. This tentative assignment will need further confirmation by orthogonal methods. In addition, a mass species with a deconvoluted mass matching that of Fc-dimer variants (e.g. +~14 Da) is also detected in this peak, but only in stressed samples. F(ab’)2 variants, including Fab’ or LC=Fd’, and oxidized F(ab’)2, were eluted before F(ab’)2 main peak and detected in all samples. Incomplete IdeS digestion product “F(ab’)2+Fc” or “LC=HC=Fd’=LC” is detected only in stressed samples implying that IdeS cleavage site may have been blocked due to either protein-protein interaction or domaindomain interaction. Two minor peaks eluted after the F(ab’)2 main peak have deconvoluted mass matching that of F(ab’)2, likely disulfide variants of F(ab’)2, but needs to be confirmed by orthogonal methods. Interestingly, a minor peak with deconvoluted mass matching that of F(ab’)2 dimer was detected only in stressed sample C.
The presence of Fc or F(ab)2 dimer and incomplete IdeS digestion product in samples B and C under denaturing condition implies covalently-linked aggregates formed via Fc-Fc, or Fab-Fab, or Fab-Fc region, respectively. If the covalent linkage to form aggregate was through disulfide bond, the aggregate is reducible and can be confirmed by further reduction of the IdeS digest. Otherwise, the aggregate is non-reducible and the peptide mapping can be applied to elucidate the nature of the covalent linkage that led to aggregate formation. It is not the intention of current study to reveal the nature of the linkage that involved in aggregate formation, and this work will be presented in another manuscript in the future. IdeS-based subunit analysis present a unique advantage compared to peptide mapping in probing protein domain-domain interactions that form aggregate, and to elucidate the reducible or non-reducible nature of the aggregate formation.
Conclusion
IdeS-based subunit analysis in conjunction with reverse phase LC-MS has shown to be a versatile method to simultaneously characterize multiple attributes such as oxidation, aglycosylation, glycosylation, glycation, C-terminal Lys-variants, N-terminal cyclization, clipped species on domain level in this study. By adopting a mild pH for digestion and reduction, IdeS-based subunit analysis demonstrated its capability of monitoring pH-labile modification such as cyclic imide that typically converted to iso-Asp and Asp during peptide mapping digestion at slightly alkaline pH. IdeS-based subunit analysis without further reduction was demonstrated to be able to elucidate domain-domain interaction in aggregate formation. However, a proper-designed peptide mapping method is needed to reveal the nature of the covalent linkage, as well as localize the amino acids that participates the covalently linked aggregate formation. It is noteworthy that detection of antibody deamidation or isomerization with a mass change of less than1 Da remains a challenge for IdeS-based subunit analysis. IdeS-based subunit analysis involves simpler and faster sample preparation, and ease of data interpretation compared to that of peptide mapping, making it an attractive rapid screening tool that leads to simultaneous characterization of multiple attributes of antibodies at domain levels.
Acknowledgements
We are indebted to Yan Wang for providing peptide mapping data of the stressed mAb1 samples, and Biogen PBC team for providing SEC aggregate fractions of mAb3 for this study.
References
- King S. The Best selling drugs of all Time; Humira joins the elite. Forbes, Jan 18, 2013
- Liu H, Gaza-Bulseco G., et al. Heterogeneity of monoclonal antibodies. J. Pharm. Sci., 2008; 97 (7): 2426-2447
- Zhang Z, Pan H and Chen X. Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spec Rev, 2009; 28: 147-176
- Rogers RS, Nightlinger NS, et. al. Development of a quantitative mass spectrometry multi-attribute method for characterization, quality control testing and disposition of biologics. mAbs 2015;7:881-90.
- Bennett KL1, Smith SV, et. al., Monitoring papain digestion of a monoclonal antibody by electrospray ionization mass spectrometry. Anal. Chem. 1997; 245:17-27.
- Jonathan S, Fredrik O and Alain B, Rapid and improved characterization of therapeutic antibodies and antibody related products using IdeS digestion and subunit analysis. (minireview) Analyst, 2016; 141:3114-3125
- An Y, Zhang Y, Mueller H.-M, Shameem M and Chen X, A new tool for monoclonal antibody analysis, Application of IdeS proteolysis in IgG domain-specific characterization, mAb, 2014; 6 (4): 879–893
- Formolo T, Ly M., et al., Determination of the NIST mAb Primary Structure, ACS Symp. Vol. 1201, 2014; Chapter 1, 1-62
- Vlasak J.; Ionescu R., Fragmentation of monoclonal antibodies, mAbs 2011; 3: 253–263.
- Wakankar AA, Borchardt RT, Eigenbrot C, Shia S, Wang YJ, Shire SJ, and Liu JL, Aspartate Isomerization in the Complementarity-Determining Regions of Two Closely Related Monoclonal Antibodies. Biochemistry 2007; 46: 1534-1544
- Oliyai C., and Borchardt RT. Chemical pathways of peptide degradation. VI. Effect of the primary sequence on the pathways of degradation of aspartyl residues in model hexapeptides, Pharm. Res. 1994;11, 751-758
- Oliyai C, and Borchardt RT Chemical pathways of peptide degradation. IV. Pathways, kinetics, and mechanism of degradation of an aspartyl residue in a model hexapeptide, Pharm.Res. 1993; 10: 95-102
- Valliere-Douglass J., et al., Separation and characterization of an IgG2 antibody containing a cyclic imide in CDR1 of light chain by hydrophobic interaction chromatography and mass spectrometry. Anal. Chem 2008;. 80: 3168-3174
- Cacia J, Keck R, Presta LG, and Frenz J. Isomerization of an aspartic acid residue in the complementarity-determining regions of a recombinant antibody to human IgE: identification and effect on binding affinity, Biochemistry 1996; 35: 1897-1903
- Eakin CM, et. al., Assessing analytical methods to monitor isoAsp formation in monoclonal antibodies, Frontiers in Pharmcology, 2014; 5:1-9
- Haverick M, Mengisen S, Shameem M. and Ambrogelly A, Separation of mAbs molecular variants by analytical hydrophobic interaction chromatography HPLC: overview and applications, mAbs, 2014; 6 (4): 852–858
Author Biographies
Dr. Li Zang is an associate director overseeing the Protein Analytical Development department of Biogen. Her Ph.D. focused on development and application of highly sensitive liquid chromatography-mass spectrometry technology for breast cancer biomarker discovery. After joining Biogen in 2005, Li developed her expertise in biopharmaceutical development especially in detailed protein structure characterization using separation and mass spectrometry. She has participated in the development of a large category of biopharmaceutical programs at Biogen over the last 12 years, including monoclonal antibodies, receptor-Fc fusion proteins; blood coagulation factors, bi-specific antibodies, antibody-drug conjugates, biosimilar programs and small endogenous proteins.
Dr. Yunyu (Linda) Yi is a Scientist II in Protein Analytical Development department of Biogen. She has developed her expertise in recombinant therapeutic protein characterization by electrophoresis, liquid chromatography, as well as LC-MS or LC-MS/MS over the last 12 years. She has participated drug development for monoclonal antibodies, blood coagulation factors, bi-specific antibodies, fusion proteins, protein vaccines, and biosimilars for preclinical and clinical programs.