High performance liquid chromatography (HPLC) is probably the most widely used analytical technique in the pharmaceutical industry for characterizing new drug substances and drug products. In recent years, the use of very high pressure liquid chromatography (VHPLC) using columns packed with sub-2 micron particles to improve chromatographic efficiency has seen increased utilization for pharmaceutical development applications to provide faster separations without significantly compromising chromatographic efficiency[1-5]. For example, runs times for impurity profiling applications which resolve all components of interest can typically be reduced from approximately 30-60 minutes to 5-10 minutes. An example of this is illustrated in Figure 1 where the chromatographic run time was decreased from 60 minutes to 8 minutes without sacrificing the chromatographic efficiency needed to resolve the impurity peaks in the sample. For in-process control applications, which may only need to resolve a couple of components, run times of less than a couple of minutes are now being achieved. As a result, scientists are now investigating developing on-line sampling techniques coupled to VHPLC systems to provide near real-time results in monitoring liquid process streams. Another significant benefit of these reduced run times is they also provide for more efficient method development and troubleshooting since a greater number of test experiments can be performed within a given workday. This represents a significant benefit to pharmaceutical development scientists who often must ensure the appropriateness of analytical methods when changes are implemented to optimize the drug substance and drug product manufacturing process.

Chromatograms of an impurity mixture analyzed using HPLC and VHPLC method conditions. The figure illustrates that the chromatographic run time was decreased from 60 minutes to 8 minutes with adequate sacrificing the chromatographic efficiency to resolve the impurity peaks in the sample.
For some more challenging separations, profiling methods may require extremely shallow gradients to separate critical components to provide adequate chromatographic resolution. In these applications VHPLC can also be used to provide more efficient, and perhaps rugged separations. In Figure 2, the chromatograms of the impurity mixture were obtained using columns packed with either 1.8 or 3.5 μm particles, but of the same column length. These chromatograms illustrate the 1.8 μm particle column provided increased resolution between the API peak and the two critical impurities which elute just before the API peak. Although no time-savings were realized between the two systems, the improved resolution may provide for more robust methods and provide greater confidence that this method could be successfully transferred to worldwide QC labs or CROs.
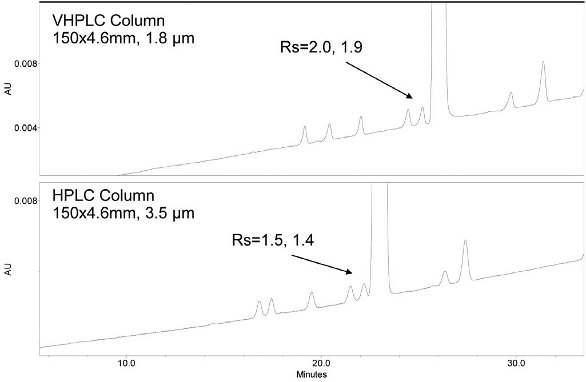
Chromatograms of an impurity mixture comparing results obtained using columns packed with 1.8 and 3.5 μm particles, otherwise similar conditions. In this application, the 1.8 um particle column provided increased chromatographic efficiency. Although no significant decrease in run time was achieved, the increased resolution may provide for increased method robustness and confidence that the method could be successfully transferred to worldwide QC labs.
Today’s pharmaceutical development organizations often leverage domestic and off-shore contract labs to perform analytical characterization, utilize chemical vendors to synthesize intermediate compounds and manufacture drug products, and often reassign existing projects within the organization to more effectively utilize available resources. As a result, pharmaceutical development scientists must also ensure methods/equipment are rugged and local QC and CRO labs are equipped and trained to run these methods. While we are currently seeing an increased availability of VHPLC instruments in QC labs around the world, the large implementation costs in terms of capital equipment budgets, time, and personnel training has still limited its widespread implementation. Many companies have resisted developing new VHPLC methods to replace lengthy (e.g. > 60 min) legacy HPLC methods due the significant costs incurred to update worldwide regulatory filings with new methodology. This cost can be significant when considering that some products may be marketed in more than 50 countries worldwide requiring a significant investment in filing costs, and also in time spent amending the existing regulatory filings. Until a time when VHPLC enjoys widespread instrument availability, pharmaceutical development scientists will need to consider mechanisms to provide comparable methods which can provide equivalent analytical results when only conventional HPLC systems are available. In the remainder of this article, we will highlight some potential factors which warrant consideration in developing VHPLC methods to ensure equivalency with conventional HPLC conditions and also to ensure equivalent profiles are obtained on different VHPLC instruments.
VHPLC Method Development
VHPLC is a natural extension of HPLC, which possesses the same chromatographic theory and principles. As a result, scientists can not only develop VHPLC methods “from scratch” for new projects and needs, but also adapt existing HPLC methods to provide more rapid or rugged VHPLC versions. There are many literature references which describe the transfer of conventional HPLC methods to VHPLC equivalent methods. Briefly, the most straightforward approach to developing VHPLC conditions for existing HPLC methods is to maintain most of the existing HPLC conditions (e.g. mobile phase composition, column chemistry, column temperature, gradient steps) and simply geometrically scale the gradient step times and injection volumes to account for the different column volumes of the HPLC and VHPLC columns[6]. Most vendors provide simple software programs to facilitate this conversion. Although a scaling approach can provide a good starting point to developing equivalent VHPLC separations, several other factors should be considered prior to finalizing the method.
To leverage the productivity enhancements of VHPLC, more labs are developing and utilizing VHPLC methods, especially when the need to transfer methods is not present. The ability to initially develop faster separations provides an obvious benefit in that a greater number of individual experiments can be performed during a workday which may provide for developing a more effective set of unattended, overnight investigations. In practice, effective method development is somewhat limited by the samples that are provided to the method development scientist. These samples should contain a suitable level and number of potential impurities to effectively guide the method development.
We have found it useful to incorporate a screening strategy to be able to screen some of important method parameters. In practice, our initial screening steps involve investigating the effect of mobile phase pH, organic composition, and column chemistry. Mobile phase pH screening is performed over a range from pH 2 to 9 to screen peak shape and retention time shifts of impurities. We then screen different columns and organic solvents to determine appropriate starting conditions for method development. Column screening has become more effective as more sub 2 μm column chemistries have become commercially available. The availability of columns with polar-embedded groups and hydrophilic interaction (HILIC) columns provide a different selectivity compared with regular C18, C8 and phenyl columns. After screening the solvent systems and columns, we then optimize other parameters such as column temperature and gradient profile screening, often using method development simulation software packages. This approach, aligned with QbD, provides a global understanding of the effects of different chromatographic parameters on the separation.
Column Considerations
Column chemistry is a major parameter which affects selectivity and therefore should be kept the same for both HPLC and VHPLC conditions. Since the silica particle properties also play an important role in column selectivity, a practical consideration for column selection is to use the same brand column (e.g. from the same manufacturer), which only differs in particle size and column dimensions to minimize selectivity differences. Selectivity changes caused by using different column chemistries could result in peak co-elution or a different impurity elution order upon method conversion. Use of the same column chemistries will typically provide similar elution profiles being observed for both method conditions. In the case where such VHPLC version columns are not available, efforts need to be made such that the overall selectivity should be as similar as possible between two columns. Currently, many vendors offer software programs which can be used to assess column equivalency.
When considering column dimensions, the theoretical efficiency of a column is proportional to the column length, and inversely proportional to particle size. Therefore, if the same column efficiency is desired for VHPLC and HPLC conditions, the column length to particle size ratio should be kept relatively the same. However, from a practical stand point, the uniformity of either the particle size or the packing bed of sub-2 μm particles is not as good as columns with 3.5 μm particles[4]. As a result, the real column efficiency for sub-2 μm columns is somewhat less than the theoretically predicted values. For example, a 10 cm long VHPLC column packed with 1.7 μm particles will theoretically provide ~30% more efficiency than a 15 cm long HPLC column packed with 3.5 μm particles. In practice however, a 10 cm long VHPLC column packed with sub-2 μm particles offers similar efficiencies to 15 cm long HPLC column packed with 3.5 μm particles. Many scientists, however, prefer to utilize 5 cm long VHPLC columns to further increase the speed of the analysis. This approach is certainly possible if the resolution between critical impurities analyzed in the separation is sufficient that a slight decrease in overall chromatographic efficiency will not compromise the separation.
Sample preparation parameters include sample concentration, diluent and extraction procedures. Since these parameters will likely have minimal effects on the VHPLC separation, keeping them the same will simplify the method validation. However, additional thought should be given to the organic content of the sample diluent if methods are being scaled down to narrow-bore VHPLC columns (e.g. <3 mm i.d), which are more readily available. Scientists could begin to experience poor peak shape of early eluting peaks and irreproducible retention due to poor sample loading onto a narrow-bore column. To prevent this, lower levels of organic solvent in the sample diluent may be required.
VHPLC Instrumentation Differences
An additional consideration for VHPLC method development is the differences between commercially available instruments. In general, the configuration of HPLC instruments has become largely standardized over the years however, differences do exist between HPLC and VHPLC equipment. In addition, with the rapid improvements in instrumentation which we are currently seeing, VHPLC instruments from different vendors are provided with somewhat different configurations, such as pressure rating, system delay volume, injector design, etc. This may lead to method robustness issues, i.e., VHPLC methods developed on a particular instrument may provide different elution profiles when run on other instruments. Scientists need to consider such differences during the method development stage to avoid future method transfer issues. For example, although quaternary systems are commercially available, most VHPLC instruments are only available as binary pumps. This factor should be considered before pursuing developing any tertiary gradients.
UV detection is the most common detection mode used in HPLC and VHPLC applications. As detection has no effect on separation, detection parameters, such as detection wavelength, should be kept the same. However, some other detector settings, such as sampling rates and filter time constant, may need to be adjusted to accommodate the narrower peak widths encountered when using VHPLC. Typically, chromatography software needs at least 10-15 points across the peak to ensure a reproducible integration. Since VHPLC peak widths are typically only a few seconds, a sampling rate of at least 5 points per second is generally required. The same consideration should also be used when applying other detection techniques, such as mass spectrometry.
Currently there are more than ten different instrument vendors who provide VHPLC systems. Pressure ratings with current VHPLC instruments from different vendors range from 9,000 to 19,000 psi, and currently, columns have different pressure ratings as well. Additionally, the range of flow rates capable of delivering backpressures greater than 5000 psi varies from vendor to vendor. Fortunately, the use of smaller particle columns provides a wider range of optimal linear flow rates since the van Deemter curves exhibit a broader optimum in the C-term region. As a result, flow rate adjustments needed to accommodate different VHPLC systems should not have a significant effect on efficiency. As a result, methods developed on a VHPLC system which provide greater than 10,000 psi backpressure, can be modified by decreasing the flow rate to be run on VHPLC systems with lower backpressure limits. Although the run times will need to be increased proportionally to the decrease in flow rate, the resolution between critical pairs of impurities should be able to be preserved.
VHPLC and HPLC systems can also be expected to have different system dwell volumes which will provide different significant delays to the elution of peaks during gradient elution. Additionally VHPLC systems from different vendors may also have different instrument dwell volumes which can also provide subtly different gradient elution profiles. In order to accommodate the differences, an appropriate isocratic hold period can be employed to the VHPLC gradient on an instrument with a smaller dwell volume. The length of this isocratic hold can be adjusted accordingly when the method is run on different instruments with different dwell volumes. Currently, several vendors offer simple software programs to determine appropriate changes needed to methods to produce similar results on with different dwell volumes. Employing dwell volume adjustments such as these to gradient methods does not require revalidation as indicated in the current USP Chapter <621> [7].
It is well known that insufficient on-line mixing of mobile phases can cause baseline disturbances when UV-absorbing additives such as trifluoroacetic acid (TFA) are used [8,9]. Pharmaceutical development scientists often face the need to utilize low detection wavelengths to detect low molecular weight intermediates, starting materials, or excipients which lack a suitable chromophore. When using low UV wavelengths with mobile phase additives, inefficient mixing can provide periodic baseline noise during gradient elution which can make it difficult to identify impurity peaks and limit sensitivity of methods unacceptably above the ICH guideline of 0.05%. This effect may be more pronounced on VHPLC systems which typically incorporate smaller volume mixers to decrease instrument dwell volume, and therefore provide less mixing efficiency. Accordingly, larger volume mixers or new mixer designs may be needed if the desired method sensitivity cannot be attained due to issues with the baseline. The effect of using larger volume mixers is shown in Figure 3. In this figure, the baseline ripple is less evident when the larger volume mixer is employed which makes facilitates identifying the peaks at the 0.1% level. Recently several new low-volume mixer designs have been introduced which are either based on multilayer microfluidics technology or conceived from studies of micro reactor technologies to deliver efficient, low-volume mixing (35 - 50 μl). In our experience, we have been very impressed with the ability of these new mixer designs to deliver stable baselines for these difficult applications[10].
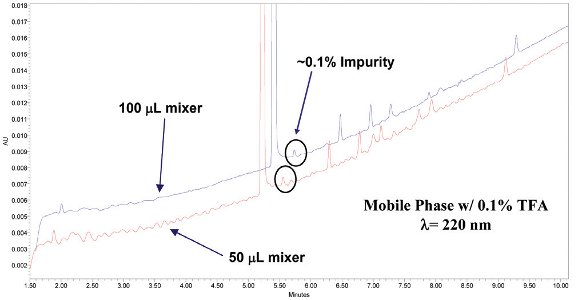
Chromatograms of an impurity mixture analyzed on a VHPLC system utilizing two different mixer volumes. Note the chromatogram obtained using a 100 μL mixer provided a more stable baseline to facilitate the identification of low level peaks in the chromatogram.
Effect of Column Temperature
Since column temperature also affects selectivity and retention, it should be kept the same for both VHPLC and HPLC conditions. However, for columns packed with sub-2 micron particles, it has been found that both longitudinal and radial temperature gradient exist within the column due to the frictional heating between the mobile phase and small particles. While the radial temperature gradient is generally not a concern for columns sitting in a forced air oven, a significant temperature increase can occur along the longitudinal direction (up to 20 ºC higher in the column outlet than inlet)[11, 12]. This longitudinal temperature gradient can cause a decrease in retention, and may also change selectivity if it is sensitive to temperature. To mitigate this effect, the column temperature in VHPLC may need to be decreased to provide equivalent separations. This effect is illustrated in the chromatograms in Figure 4. At a 0.5 mL/min flow rate, the impurity in front of the main peak was well separated using a column temperature of 55ºC. However,increasing the flow rate to 1.0 mL/min caused the impurity to co-elute with the main peak. However, separation of the impurity and the main peak was restored by reducing the column temperature to ~ 40 ºC when using the higher flow rate. Based on the structures of the impurity and main peak, the change in selectivity observed in the figure may be due to the increased temperature affecting of the pKa of the one or both of the compounds.
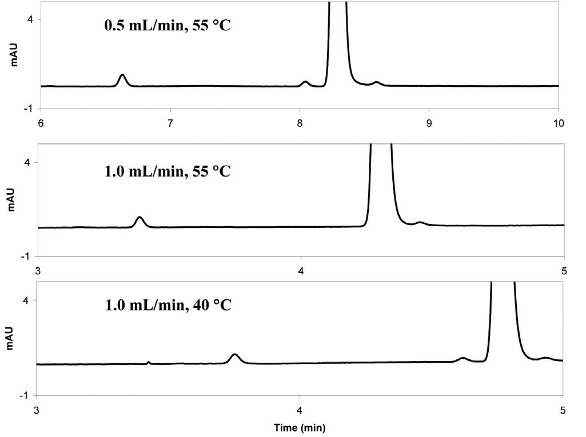
Figure 4: VHPLC chromatograms illustrating the effect of a longitudinal temperature gradient may have on retention. At a 0.5 mL/min flow rate, the impurity in front of the main peak was well separated using a column temperature of 55 ºC. However, increasing the flow rate to 1.0 mL/min caused the impurity to co-elute with the main peak. Adequate separation was achieved by reducing the column temperature to ~ 40 ºC.
Summary
Today’s pharmaceutical development organizations often leverage domestic and off-shore contract labs to perform analytical characterization, utilize chemical vendors to synthesize intermediate compounds and manufacture drug products, and often reassign existing projects within the organization to more effectively utilize available resources. As a result, scientists often need to ensure that methods can be successfully implemented in different labs throughout the development lifecycle. Until a time when VHPLC enjoys widespread deployment, pharmaceutical development scientists will need to consider strategies to provide comparable methods which can provide equivalent analytical results when only conventional HPLC systems are available.
To address this issue, it may be practical to provide equivalent VHPLC and HPLC methods and allow either method to be used for analytical testing. The VHPLC version can be transferred to the labs where such instruments are available; otherwise the HPLC version can be transferred. An interesting alternative to the VHPLC approach presented in this article being investigated by several labs is the use of fused-core (superficially porous) column particles which allow very fast separations of small molecules at lower backpressures typically available in most conventional HPLC instruments[13]. APR
References
- N. Wu, A. M. Clausen, J. Sep. Sci. 2007, 30, 1167.
- A. D. Jerkovich, R. LoBrutto, R. V. Vivilecchia, LCGC North Am. 2005, 15.
- M. W. Dong, LCGC North Am. FIELD 2007, 89.
- A. De Villiers, F. Lestremau, R. Szucs, S. Gelebart, F. David, P. Sandra, J. Chromatogr., A 2006, 1127, 60.
- A. D. Jerkovich, J. S. Mellors, J. W. Jorgenson, LCGC North Am. 2003, 21, 600.
- B.L. Kleintop, Q. Wang, in “Characterization of Impurities and Degradants Using Mass Spectrometry” M.S. Lee, G. Chen, eds., in press.
- US Pharmacopeia, USP32-NF27, General Chapter 621, 2009.
- H. E. Schwartz, B. L. Karger, P. Kucera, Anal. Chem. 1983, 55, 1752.
- K. Choikhet, B. Glatz, G. Rozing, LC-GC Eur. 2003, 16, 2.
- B.L. Kleintop, Q. Wang, Presented at the 2010 Pittsburgh Conference on Analytical Chemistry and Applied Spectroscopy, Orlando, FL, 2010.
- A. de Villiers, H. Lauer, R. Szucs, S. Goodall, P. Sandra, J. Chromatogr. A 2006, 1113, 84.
- F. Gritti, G. Guiochon, Anal. Chem. 2008, 80, 5009.
- DeStefano, J.J., Langlois, T.J., Kirkland, J.J., J. Chrom. Sci, 2008, 46, 254.
Brent L. Kleintop, Ph.D. is currently an Associate Director within Analytical R&D at Bristol-Myers Squibb and has been working within the pharmaceutical industry for more than 15 years. Brent’s scientific interests center upon improving the efficiency of analytical chemists through application of modern analytical instrumentation. Readers may contact him directly at: [email protected]
Qinggang Wang, Ph.D. is currently a Senior Research Investigator within Analytical R&D at Bristol-Myers Squibb, where he has worked for the past 7 years. Quinggang’s research interests involve utilizing different separations-based methods to improve characterization of drug substances and drug products.