Introduction
Controlled onset oral drug delivery can be simply described as drug release following a pre-determined time delay; the challenge for the formulation scientist is achieving this reliably in vivo. Some of the key life cycle management targets for this technology include engineering drug delivery to meet the challenge of nocturnal and pre-wakeup drug delivery for diseases such as rheumatoid arthritis and cardiovascular conditions, which typically exhibit a circadian morning exacerbation of symptoms [1,2], facilitating daytime repeat dosages and providing pharmacological opportunities for one or more different drugs to be delivered at different times or rates from a single dosage unit.
The time taken for an oral dose to transit through the gastrointestinal tract (GIT) will vary between and even within individuals, and is further affected by factors such as whether the patient has eaten [3], what he/she has eaten [4], and whether he/she is awake or asleep [5]. Thus, release of drug from the onset-controlled formulation following ingestion should be entirely dependent on the programmed time delay, irrespective of its location in the GIT or the physiological environment.

As an oral dosage form travels along the GIT, it will be exposed to a changing physiological environment, with pH varying between 1.2 and 7.5, exposure to digestive juices and enzymes, and on arrival in the lower bowel, a gradually decreasing availability of water. In addition, the mechanical forces exerted on the dosage form by the GIT will vary, from the milling and grinding experienced in the antrum of the stomach in the fed state, to the mixing and propulsive contractions in the intestines, with random periods of stasis. The forces exerted on a formulation traveling through the gut have been estimated to range from 1.2N in the fasted stomach and intestine to 1.9N in the fed stomach [6]. In addition, the physical effect of the presence of food should not be underestimated, both in terms of a physical abrasive effect on the dosage form, and the potential for a component of the food to interact with it [7].
Therefore, since we cannot predict the precise location of the dosage form at the time of drug release, the performance of a successful formulation must be independent of these patient-specific variables.
A range of different formulation approaches with the objective of achieving this aim have been reported in the literature.
Multi-component Tablets
Perhaps the most widely cited example of controlled onset delivery is a variation of an osmotic formulation which, in its original format, was a tablet comprising a swellable osmotic “push” layer situated below the drug-containing layer. The bilayer tablet was surrounded by a hydrophilic delay barrier coating and an outer semi-permeable membrane. As water slowly penetrates into the system through the outer rate controlling layers, the osmotic component swells, forcing the drug out through a precision laser drilled orifice. This controlled-release oral delivery technology was modified by the addition of a hydrophilic polymer layer that caused a time delay prior to water reaching the swellable osmotic “push” layer, and was supplied as a formulation for controlled onset release of verapamil. This formulation was designed to be taken at bedtime by hypertensive patients, enabling a 4-5 hour delay before release of the active ingredient, to target the known early morning rise in blood pressure/heart rate.
While not strictly falling into the category of a tablet in the conventional sense, an injection molded drug delivery system provides an alternative means of delivering controlled onset of drug release, and variations on the basic design are currently at different stages in the development process [8]. The device consists of a non-permeable injection-molded polymer outer case that forms an elliptical cylinder with openings at either end. Because the surface area for erosion of excipients from these openings remains constant, the drug and excipients can then be layered and arranged within the impermeable shell, to control the time and rate of drug release.
Press-coated Tablets
 Figure 1. Cross-section of a typical “tablet within a tablet” formulation.
Figure 1. Cross-section of a typical “tablet within a tablet” formulation.Press coating is a method of producing “tablet within a tablet” formulations (Figure 1), where the outer press coated layer is generally designed to form a barrier to water ingress and hence provide a time delay prior to drug release. This type of formulation is manufactured using commercially available tablet presses that integrate core tablet production with compression coating of a dry powder outer layer.
The pulsed release system described by Pozzi [9] is an example of this approach, where an outer press coated layer composed of a mixture of hydrophobic and surfactant components provides an erodible barrier, preventing drug release from the core tablet for a period of time which is determined by its thickness. In vivo studies of this formulation using gamma scintigraphy to monitor the behavior of the formulation in combination with pharmacokinetic determination of salbutamol in plasma demonstrated that three- and six-hour delay periods could be achieved using this formulation [9].
Sangalli et al. [10] presented a similar system in which the barrier coating was composed of a gel-forming hydrophilic polymer layer, and demonstrated that the thickness of the layer can be used to control the lag time before sustained drug release through the gelled hydroxypropyl methylcellulose (HPMC) matrix [10,11].
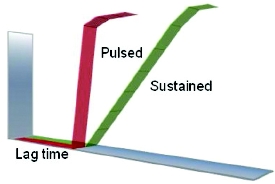 Figure 2. Modulation of release profi le from core tablet using timedelayed controlled onset technology.
Figure 2. Modulation of release profi le from core tablet using timedelayed controlled onset technology.A platform for time-delayed controlled onset technology [12] is tuneable to achieve a range of different controlled onset release profiles tailored to a particular therapeutic strategy. Essentially, the formulation is composed of a drug-containing core tablet surrounded by an erodible compression coating. The controlled erosion of the coating creates a time delay before the core tablet is exposed to the surrounding media, ultimately enabling release of the drug contained in the core. The time of onset of drug release is then further controlled by the nature and ratio of hydrophilic/hydrophobic materials in the coating and the thickness of this layer. The multi-component structure of this formulation offers the potential to manipulate the drug release profile, through modulation of the release behavior from the core tablet (Figure 2). Further opportunities exist to extend the applications of this system by the addition of an enteric coat around the formulation, in order to delay the “release countdown” until after the tablet has emptied from the stomach. By addition of a barrier layer that is programmed to gradually erode over the relatively well-documented transit time through the small intestine (around 3-4 hours), targeted delivery of drug to the colon can be realized.
As previously outlined, the local physiological environment to which a controlled onset formulation is exposed can be highly variable, and the exact location of release remains unpredictable. Crucially, this platform formulation has been shown to be independent of pH, agitation, and fat content in vitro [13]. This is an essential factor in creating a robust formulation that behaves as independently of patient specific factors as possible, to achieve reproducible clinical outcomes. The difficulty in predicting in vivo behavior from standard USP dissolution methods is well known, and it is difficult, time consuming, and often expensive to even attempt to replicate the complexities of the human GIT in vitro. Wherever possible, carrying out in vivo studies is essential to confirm the robustness of formulation performance, particularly in controlled onset formulations where release must be independent of local GIT conditions. Due to this variability in site of drug release, pharmacokinetic profiles obtained from controlled onset formulations may be difficult to interpret; therefore, employing an in vivo visualization technique to determine site of drug release enables ultimate confidence in product performance.
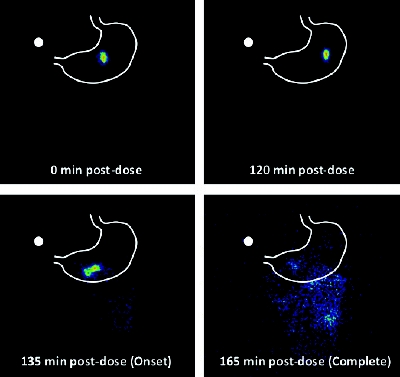 Figure 3. Scintigraphic images of 99mTc-labeled delayed release tablet demonstrating onset of release 2.25 hours after administration.
Figure 3. Scintigraphic images of 99mTc-labeled delayed release tablet demonstrating onset of release 2.25 hours after administration.Gamma pharmacoscintigraphy is a technique in which a formulation is labeled with a radioactive isotope, such as 99mTc or 111In, which enables visualization and quantification of the dosage form in the gut after ingestion by the patient, thus allowing location to be correlated with simultaneously obtained pharmacokinetic data. Pharmacoscintigraphy has been used to confirm the robust in vivo performance of the platform technology described above for therapeutic applications such as sleep maintenance induction [14], and the early morning pain and stiffness of rheumatoid arthritis [13]. Figure 3 shows the observation of radiolabel release at 2.25 hours from the core tablet containing a drug for sleep maintenance induction in vivo using gamma scintigraphy in volunteers [14], which correlates well with the in vitro lag time of 2.5 hours (data not shown).
Ref. 15 describes an oral drug delivery formulation that also consists of a drug-containing core surrounded by a controllable delay layer, in this case composed of hydrophobic wax and brittle material [15]. On exposure to water, the balance of excipients controls the rate of water ingress through the barrier coating, until subsequent swelling of the core mechanically weakens the barrier, allowing rupture and delayed burst release of core contents. This formulation has achieved clinical success in the CAPRA-2 study [16], and a formulation has been developed that recently received approval in the U.S.A. for delivery of low-dose prednisolone for chronotherapeutic treatment of early morning pain and stiffness in rheumatoid arthritis.
Capsule Formulations
An alternative approach to tablet formulations is the use of a capsule platform from which to control the onset of drug release. In a prime example of this approach [17], drug release was delayed by coating a gelatin capsule body with an impermeable layer composed of a polymer such as ethylcellulose, or constructing the capsule body itself from molded plastic. The contents of the capsule (active pharmaceutical ingredient [API] and an expulsion excipient) were then sealed in the capsule body using a hydrogel plug composed of cross-linked polyethylene glycol (PEG). On contact with gastric fluids, the hydrogel plug swells in a controlled manner, developing a frustro-conical shape as it gradually pulls itself out of the rigid capsule body, finally allowing the drug contained within to be released. The length of time for this process can be refined by changing the length of the hydrogel plug, or altering the depth to which it was inserted. The controlled onset performance of this system was found to be independent of the nature and pH of the medium [18], and its in vivo performance in humans has been reported in the literature [18,19].
Since the hydrogel plug was the sole determinant of the time at which drug was released from the above capsule formulation, one of the key technical constraints was the precision required in the placement of the plug within the capsule during manufacture. A strategy to overcome this difficulty was reported by McConville et al., where the “moving” part of the hydrogel plug was replaced by a slowly eroding tablet plug. The erosion mechanism meant that onset of drug release was controlled exclusively by the composition and thicknesses of the plug, since there were no “moving parts” in the system. The role of the erodible plug in controlling the onset of drug release was confirmed in vitro for this formulation [20–22]. Subsequent pharmacoscintigraphic studies confirmed this role in vivo, while also demonstrating that the system performed more robustly in the upper GI tract, since lack of availability of water in the distal gut was hypothesized to have slowed erosion for the extended lag time system [23]. This further emphasizes the desirability of designing controlled onset formulations in such a way as to ensure that performance is as independent of local gut conditions as possible.
Pellet Formulations
An approach that has found some success as an alternative to the formulation of monolithic dosage forms is the production of multi-layered pellets to control the onset of drug release. A drug-containing core (or inert core surrounded by a drug layer) is surrounded by one or more layers of excipients that are designed to control the onset of drug release, and in this case conventional pharmaceutical coating equipment is generally used to create the multi-layered pellets. The pellets are normally then delivered in a hard gelatin capsule. One advantage of this method is the relative ease with which different pellet populations can be combined, producing a complex release profile.
An early example of this approach is the Time Controlled Explosion System (TES) reported by Ueda [24,25], wherein a core composed of drug layered on a nonpareil seed is surrounded by a layer of the swelling excipient low-substituted hydroxypropyl cellulose (L-HPC), with a final outer coating of the water-insoluble polymer ethylcellulose. The brittle outer membrane allows water to slowly penetrate into the core, causing gradual swelling of the L-HPC until the outer membrane ruptures, releasing the drug. The time of this burst release could be controlled by varying the thickness of the outer membrane.
A drug delivery technology based on chronotherapeutic oral drug absorption consists of drug-containing pellets that are coated with a combination of water-soluble and insoluble polymers. On exposure to water, the soluble polymer gradually dissolves away, leaving pores in the remaining water insoluble polymer through which drug can diffuse, controlling the time and rate of release. This technology is currently used in a formulation that is taken at bedtime to deliver verapamil at around 4-5 hours post-dose [26].
A multiparticulate bead system is composed of drug layered onto an inert core, further surrounded by one or more layers of release controlling polymer [27]. This technology has been used to develop propranolol hydrochloride capsules, which are taken at bedtime to produce a plasma Cmax prior to waking up, by means of a 4-hour delay before drug release.
Conclusions
Controlled onset drug delivery has a number of therapeutic clinical applications. With skillful manipulation of the excipients and construction of the system, controlled onset drug delivery systems can be manufactured relatively easily using conventional pharmaceutical equipment. With careful selection of the therapeutic entity and alignment with the specific nature of an individual disease state, these technologies can be an effective tool in meeting a clear clinical need.
References
- I. C. Kowanko, M. S. Knapp, R. Pownall, and A. J. Swannell. Domiciliary self-measurement in the rheumatoid arthritis and the demonstration of circadian rhythmicity. Annals of the rheumatic diseases, vol. 41, no. 5, pp. 453–5, Oct. 1982.
- J. E. Muller, P. H. Stone, Z. G. Turi, J. D. Rutherford, C. A. Czeisler, C. Parker, W. K. Poole, E. Passamani, R. Roberts, and T. Robertson. Circadian variation in the frequency of onset of acute myocardial infarction. The New England journal of medicine, vol. 313, no. 21, pp. 1315–22, Nov. 1985.
- F. Podczeck, C. L. Mitchell, J. M. Newton, D. Evans, and M. B. Short. The gastric emptying of food as measured by gamma-scintigraphy and electrical impedance tomography (EIT) and its influence on the gastric emptying of tablets of different dimensions. Journal of pharmacy and pharmacology, vol. 59, no. 11, pp. 1527–36, Nov. 2007.
- S. S. Davis, J. G. Hardy, M. J. Taylor, D. R. Whalley, and C. G. Wilson. A comparative study of the gastrointestinal transit of a pellet and tablet formulation. International journal of pharmaceutics, vol. 21, pp. 167–177, 1984.
- A. J. Coupe, S. S. Davis, D. F. Evans, and I. R. Wilding. The effect of sleep on the gastrointestinal transit of pharmaceutical dosage forms. International journal of pharmaceutics, vol. 78, no. 1–3, pp. 69–76, Jan. 1992.
- M. Kamba, Y. Seta, A. Kusai, M. Ikeda, and K. Nishimura. A unique dosage form to evaluate the mechanical destructive force in the gastrointestinal tract. International journal of pharmaceutics, vol. 208, no. 1–2, pp. 61–70, Nov. 2000.
- B. Abrahamsson, T. Albery, A. Eriksson, I. Gustafsson, and M. Sjöberg. Food effects on tablet disintegration. European journal of pharmaceutical sciences: official journal of the European Federation for Pharmaceutical Sciences, vol. 22, no. 2–3, pp. 165–72, Jun. 2004.
- Egalet [Online]. Available: http://egalet.com/index.php/products-and-pipeline/pipeline/. [Accessed: 01-May-2013].
- F. Pozzi, P. Furlani, A. Gazzaniga, S. Davis, and I. Wilding. The TIME CLOCK system: a new oral dosage form for fast and complete release of drug after a predetermined lag time. Journal of controlled release, vol. 31, no. 1, pp. 99–108, Aug. 1994.
- M. E. Sangalli, A. Maroni, C. Buscetti, L. Zema, F. Giordano, and A. Gazzaniga. In vitro and in vivo evaluation of oral systems for time and site specific delivery of drugs (Chronotopic technology). Bollettino chimico farmaceutico, vol. 138, no. 3, pp. 68–73, 1999.
- A. Gazzaniga, M. E. Sangalli, and F. Giordano. Oral Chronotopic drug-delivery systems— achievement of time and or site-specificity. European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik e.V, vol. 40, no. 4, pp. 246–250, 1994.
- Drug Delivery International [Online]. Available: http://www.drugdeliveryinternational.com/ licensing-opportunities/. [Accessed: 01-May-2013].
- H. N. E. Mullen, A. B. Eccleston, S. A. Hodges, L. A. Ronaldson, V. A. Sime, K. Pecek, and D. Stevens. Controlled-release diclofenac for early morning pain and stiffness. AAPS journal, vol. 13, no. S2, p. W5125, 2011.
- H. N. E. Mullen, A. B. Eccleston, S. A. Hodges, L. A. Ronaldson, V. A. Sime, K. Pecek, and D. Stevens. Correlating Formulation Behaviour with Physiological Effects for a Time-Delayed Sleep Tablet. in Proceed. Intern. Symp. Control. Rel. Bioact. Mater., 2011, p. 250.
- SkyePharma [Online]. Available: http://www.skyepharma.com/Technology/Oral_ Technology/Geoclock/Default.aspx?id=61. [Accessed: 01-May-2013].
- F. Buttgereit, D. Mehta, J. Kirwan, J. Szechinski, M. Boers, R. E. Alten, J. Supronik, I. Szombati, U. Romer, S. Witte, and K. G. Saag. Low-dose prednisone chronotherapy for rheumatoid arthritis: a randomised clinical trial (CAPRA-2). Annals of the rheumatic diseases, vol. 72, no. 2, pp. 204–10, Feb. 2013.
- M. E. McNeill, A. Rashid, and H. N. E. Stevens. Dispensing Device. U.S. Patent GB 22304421993.
- J. Binns, H. N. E. Stevens, J. McEwen, G. Pritchard, F. M. Brewer, A. Clarke, E. S. Johnson, and I. McMillan. The tolerability of multiple oral doses of PulsincapTM capsules in healthy volunteers. Journal of controlled release, vol. 38, no. 2–3, pp. 151–158, Feb. 1996.
- H. N. Stevens, C. G. Wilson, P. G. Welling, M. Bakhshaee, J. S. Binns, A. C. Perkins, M. Frier, E. P. Blackshaw, M. W. Frame, D. J. Nichols, M. J. Humphrey, and S. R. Wicks. Evaluation of PulsincapTM to provide regional delivery of dofetilide to the human GI tract. International journal of pharmaceutics, vol. 236, no. 1–2, pp. 27–34, Apr. 2002.
- A. C. Ross, R. J. Macrae, M. Walther, and H. N. E. Stevens. Chronopharmaceutical Drug Delivery from a Pulsatile Capsule Device based on Programmable Erosion. Journal of pharmacy and pharmacology, vol. 52, no. 8, pp. 903–909, Aug. 2000.
- J. T. McConville, A. C. Ross, A. R. Chambers, G. Smith, A. J. Florence, and H. N. E. Stevens. The effect of wet granulation on the erosion behaviour of an HPMC-lactose tablet, used as a rate-controlling component in a pulsatile drug delivery capsule formulation. European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik e.V, vol. 57, no. 3, pp. 541–9, May 2004.
- J. T. McConville, A. C. Ross, A. J. Florence, and H. N. E. Stevens. Erosion characteristics of an erodible tablet incorporated in a time-delayed capsule device. Drug development and industrial pharmacy, vol. 31, no. 1, pp. 79–89, Jan. 2005.
- J. T. McConville, L.-A. Hodges, T. Jones, J. P. Band, B. O’Mahony, B. Lindsay, A. C. Ross, A. J. Florence, A. J. Stanley, M. J. Humphrey, C. G. Wilson, and H. N. E. Stevens. A pharmacoscintigraphic study of three time-delayed capsule formulations in healthy male volunteers. Journal of pharmaceutical sciences, vol. 98, no. 11, pp. 4251–63, Nov. 2009.
- S. Ueda, R. Ibuki, A. Kawamura, S. Murata, T. Takahashi, S. Kimura, and T. Hata. Development of a novel drug delivery system, time-controlled explosion system (TES). IV. In vivo drug release behavior. Journal of drug targeting, vol. 2, no. 2, pp. 133–40, Jan. 1994.
- S. Ueda, T. Hata, S. Asakura, H. Yamaguchi, M. Kotani, and Y. Ueda. Development of a novel drug release system, time-controlled explosion system (TES). I. Concept and design. Journal of drug targeting, vol. 2, no. 1, pp. 35–44, Jan. 1994.
- D. Smith. A new chronotherapeutic oral drug absorption system for verapamil optimizes blood pressure control in the morning. American journal of hypertension, vol. 14, no. 1, pp. 14–19, Jan. 2001.
- Aptalis [Online]. Available: http://www.aptalispharmaceuticaltechnologies.com/content/ technology-diffucaps#tabs1. [Accessed: 01-May-2013].
Author Biographies
Howard Stevens, Ph.D., is a pharmacist whose career spans four decades of management of industrial research and development in the UK and Europe in large multinationals and small drug delivery companies. He has an extensive scientific and patent publication record, has been involved in the formation of several successful spin-out businesses and served as the Pfizer Professor of Drug Delivery at the University of Strathclyde from 1998-2009.
Fiona MacDougal, Ph.D., is a pharmacist with over 10 years’ experience in formulation development and evaluation, both in vitro and in clinical proof of concept studies. She completed her Ph.D. in Formulation Science at the University of Strathclyde in 2003, where she continued an academic role in Pharmaceutics before taking up her current position.