There are numerous tools and technologies for formulation scientists to develop the optimal dosage form for a drug. As with many aspects of science, simply having the tools on hand is not enough. Experience plays a crucial part in the development of a drug, interpretation of data, and application of knowledge appropriately to each challenge that arises to ensure progress through developmental phases.
But where does one start? Every molecule is unique in its challenges and hurdles. An experienced formulation scientist knows that there will be a number of characteristics of a molecule that determine the path of development, and whether an oral dosage form is appropriate. Even without access to physical API supply, physicochemical properties such as the molecule’s solubility, permeability, and stability can be predicted early on in the development process.
It is at this point however, where the ideal scientific solution meets real-world realities. In silico predicted characteristics may significantly diverge from experimental results. There is unlikely to be an ample supply of API material for the formulation team to run every test that they would wish to, and explore every possibility in terms of drug form and appropriate delivery system that a patient can tolerate. These two fundamental decisions however, can have a massive impact on the speed and progression of a development program.
Understanding and Optimizing the Drug Molecule’s Physicochemical Characteristics
The properties of an API are fixed as soon as its nature, polymorphic form, and manufacturing process are finalized, and so, deciding on whether the API will be a free acid, free base, or a salt, and what polymorph it will be, requires proper physical characterization and optimization of the API manufacturing process.
With limited quantities of API generally available at early stages, scientists must be strategic with limited data in making decisions. The crucial aspect is seeking the best possible answers with the limited options available, so that data can be generated to address the most common reasons for drug development attrition (poor pharmacokinetics, toxicity, and poor efficacy) as early as possible. For smaller companies who may not have in-house experience of every aspect of drug development, partnering with companies that do, and have access to equipment and resources to fully evaluate a molecule’s potential, can be highly beneficial to ensure the success of a product.
A rational starting point would be to screen a molecule to determine and quantify the most common parameters that affect the drug molecule’s behavior in the final dosage form. These include confirmation of its physical form, purity, solubility, stability, hygroscopicity, melting point, particle size, and excipient compatibility.
With this initial data, a “wish-list” of follow-up tests can start to be built up so that evidence of other potential issues, and further data can build up a drug molecule’s profile to assist in which development strategies are either feasible or not feasible. These can then be carried out as and when additional material may become available.
One of the greatest challenges for formulators is having drugs that cannot dissolve when dosed to a patient and then permeate across the gut wall into the bloodstream. Over 90% of molecules in development currently do not have adequate solubility and permeability to achieve good bioavailability.1 It is, therefore, logical to determine these properties early on, and then look at how solubility enhancing technologies can assist in overcoming any problems identified.
It is important to remember however, that an API’s properties are determined by its structure and form. Despite the temptation that may exist to try to apply an approach that worked for a different molecule in the hope that things will work exactly as they did before, the ideal methodology to employ is to start a project with an open mind and begin afresh for each new molecule.
During API development, molecule engineering can improve the solubility and stability of a drug molecule by tweaking chemical structure, potential salt forms, co-crystals, polymorphs and purity.
The challenges of candidate selection are very difficult, as there may be multiple molecule candidates to choose from, and the traditional methods to detect and generate salt forms, co-crystals, and polymorphs require significant time and API. High throughput screening and predictive in silico modelling can be employed to cover a large number of samples, although care must be taken in interpreting data from small-scale experiments and models.
If a selected API candidate is poorly soluble or unstable and is easily ionizable, then the creation of a salt form is one of the easiest options to improve one or more of these properties. Commonly adopted salt forms include hydrochlorides, tartrates, succinates, and acetates. However for drug molecules that are not easily ionizable, cocrystallization is an alternative strategy.
Co-crystals are non-ionic supramolecular complexes between the drug molecule and a co-former. There are a number of established coformers that can provide a starting point for a screening campaign, and, used successfully, can improve a molecule’s chemical stability, hygroscopicity, solubility, dissolution rate, and bioavailability.
Whether or not the selected API is developed in its pure form, or as a salt or co-crystal, the FDA and International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) require that all polymorphic crystal forms of the API are identified and characterized. Polymorphs are crystals with the same chemical composition, but possess different lattice structures and/or different molecular conformations and come about due to differences in the manufacturing process of an API. One of the most common causes of polymorphic differences is the choice of the final solvent used in the crystallization of an API. Polymorphs have different physicochemical properties (such as melting point and solubility) and each will behave differently. Although one polymorph may have higher solubility, it is likely to be metastable and will convert to the thermodynamically stable form over a short time. It is crucial that the best polymorph is selected to progress, and that this can be formed every time during manufacture and remain stable during storage.
Determining the Formulation
Once the API’s properties are fixed, these can now be used to determine the optimal drug delivery technology. The Biopharmaceutics Classification System (BCS) has been adapted to create a more delivery relevant Developability Classification System (DCS) which can be used as a very useful starting point to this process.2
The DCS classifies the molecule based on its unique dose-solubility ratio and effective permeability properties and plots it onto a chart (see Figure 1). The API’s position gives a guide as to the most likely technology to give a successful oral product. As more data are generated, this can be updated and refined, but these two initial parameters give a very good starting point for a formulator.
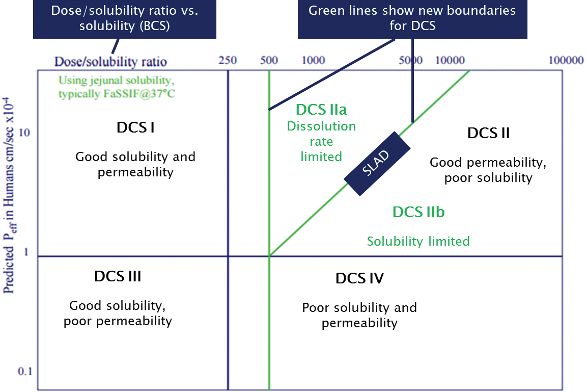 Figure 1.
Figure 1.Class I molecules (good solubility and permeability) will be relatively straightforward to develop and lend themselves towards delivery by a solution, suspension, tablet, capsule and even injectable. Patient preference and cost-effectiveness will have the major influence in final dose form; however, there may be other factors such as incompatibility with particular technologies that may narrow the choice.
Class III molecules, which exhibit good solubility but poor permeability, may need permeation enhancement technologies, but are likely to be amenable to suitably designed tablets or capsules. Those in Class IV may be acceptable in a formulation that overcomes the molecule’s poor solubility and permeability characteristics; however, in these cases, a target bioavailability of 100% is probably unrealistic. It may well be that a different candidate needs to be evaluated with better properties, or a prodrug strategy can be employed.
DCS Class II is by far the most common category for new chemical entities to fall into, and these show good permeability, but have poor solubility. 70% of drugs in development are classified within DCS Class II, but one of the many advantages of the DCS over the BCS is that this class is further divided into sub-categories, with the distinction between Class IIa and Class IIb based upon a molecule’s solubility limited absorbable dose (SLAD), below which all the dose could dissolve, and above which the fraction dissolved will diminish with increasing dose.
In DCS Class IIa, the solubility limited bioavailability results from a limited dissolution rate rather than the actual solubility itself, and for the formulator, simple particle size reduction or solutions can be highly beneficial to determine optimal drug delivery technology. For DCS Class IIb, the full dose is not expected to dissolve before exiting the small intestine, so a lipid-based solution or amorphous solid dispersion is normally a better choice for development. For molecules close to the SLAD boundary on the IIb side, co-micronization with a suitable surfactant is a strategy that can be beneficial.
Translation to In Vivo Studies
Once a number of formulation options are under consideration, evaluation of the behaviors under in vivo conditions is the next step. These tests in animals can then be used to support pharmacokinetic, toxicology, and efficacy decisions as whether to progress a drug to Phase I first-in-human clinical trials.
Conclusion
With the increased amount of knowledge, experience and screening available to formulators, many of the “predictable” pharmacokinetic issues that can increase the risk of failure early on in clinical development can be overcome. Even with limited quantities of API, a combination of in silico screening, high-throughput screening, and parallel technology evaluation can ensure data can be generated to put a molecule on a development path that has the potential for success – and more crucially, reduces the chances of failure on a path that is unlikely to ever succeed. Instead of selecting a technology without access to data, and trying to get a molecule to work within its constraints, using data to steer a molecule towards a technology that shows the greatest potential, can save precious time and resources within a project.
As more molecules are screened, and models refined to incorporate more real-life data, predictions will become more accurate to assist in drug development in the future. Debate will continue as to whether predictions will ever be 100% accurate, but experience and accumulation of data will assist, and in the absence of unlimited – and in many cases even ideal – time and resources, using models and predictive tools will provide a starting point to development programs.
Data will always need interpretation, and the skill of the scientist can never be overlooked in this. By approaching each project with an open mind, and evaluating it from a data-driven, fact-based point of view, each molecule with its own individual characteristics and inherent challenges can have the greatest chance of success.
References
- Butler JM, Dressman JB. The developability classification system: application of biopharmaceutics concepts to formulation development. J Pharm Sci. 2010; 99: 4940-54.
- Hauss, David “Oral Lipid-based Formulations: Addressing an Urgent Industrial Need”, New Jersey Centre for Biomaterials www.njbiomaterials.org/NJCB_Files/File/Skin%20 Workshop/5.%20Hauss_Presentation.pdf.