Viral contaminations of biopharmaceutical manufacturing cell culture facilities are a significant threat to product manufacture and supply and one for which having a risk mitigation strategy is highly desirable. Although they are rare, they still occur sporadically, and they have a potential to incur serious consequences such as plant shutdown, manufacturing interruption, cost associated with investigation and remediation, business impact, drug supply interruption, regulatory impact, and patient safety, to mention a few. Introduction of adventitious agents can occur at any step in a complex manufacturing process of biologics when cells are exposed to raw materials, and in many cases they may only be detected after amplification by growth in the cell substrate or in production bioreactors [1,2]. The most recent contaminations in mammalian cell culture have been associated with the raw materials; therefore it is becoming increasingly critical to address the potential entry of different contaminants into the process and identify remediation plans in order to mitigate manufacturing downtime, the risk to patient supply, as well as the overall production costs of a contamination [3,4]. Events of viral contamination due to raw materials, especially bovine serum and porcine trypsin, have been reported; some examples are presented in Table 1.
Table 1. Selected published instances of Contamination in the production of Biologics

Viral contamination of mammalian cell cultures used for biopharmaceutical manufacturing of monoclonal antibodies, therapeutic recombinant proteins, and vaccines has led to requirements by regulatory authorities for manufacturers to develop risk mitigation strategies. It is widely acknowledged that testing alone is not sufficient to assure safety from any adventitious agent contamination, and additional measures are needed at various stages from raw material supply to the manufacture of the finished product. In addition to the virus contamination risk, mammalian cell culture processes are inherently prone to contamination by microorganisms that can pass through 0.1-μm pore size filters such as Leptospira licerasiae, which have been associated with a recent contamination event [5].
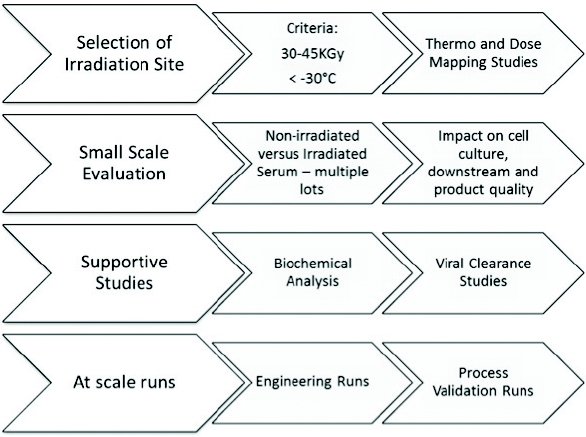
As a part of continuous improvement efforts to eliminate potential risk for adventitious agent contaminations, Genzyme and Sanofi Pasteur, A Sanofi Company, and the Vaccines Division of Sanofi , respectively, have initiated a program to further mitigate the risk from adventitious agent contamination arising from the use of raw materials, comprising the following: selection and evaluation of raw materials and their source and suppliers, specification review and upgrade, replacement/ removal/refinement (3Rs) of raw materials, as well as evaluation of clearance technologies such as gamma irradiation, high-temperature short-time treatment (HTST), nanofiltration, and/or ultraviolet-C (UVC) treatment methods as a barrier approach to prevent contamination of mammalian cell-based operations and final products. Furthermore, extensive characterization and testing of the cell substrate, lot-to-lot release testing for adventitious contaminants, raw material selection and testing, and viral clearance demonstration of the downstream purification unit operations are embedded to this integrated approach. However, the impact of implementing new treatment technologies and introducing new/replacing existing raw materials on the cell growth, productivity, and product quality cannot be predicted; therefore comprehensive assessments using small-scale studies are required to assess their suitability. The purpose of this article is to recommend a holistic biosafety approach in an effort to mitigate the risk of viral contamination associated with raw materials.
Raw Materials Supply Quality Management: Reducing Risk by Ensuring Quality and Availability
Raw material traceability is generally considered suboptimal, as a recent case of adulterated fetal bovine serum has highlighted. It is therefore very important to institute an effective supplier management program as a key prerequisite to ensuring the quality, safety, traceability, and general compliance of given raw materials. Items to consider when selecting a new vendor include its quality systems and solvency, as well as its length of time in business, geographic area, and whether it supplies multiple industries. Furthermore, ensuring both the availability and the qualification of secondary suppliers is essential. Supplier quality and technical agreements should include strict change control sections as well as details on management of vendor notification changes in its product or suppliers, including the impact assessments from both supplier and manufacturer in the event the supplier would eventually introduce any change.
In this respect, risk assessments are an important tool for ensuring the safety, efficacy, consistency, and supply of pharmaceutical products based on ICH Q9 guideline.
Throughout the risk assessments, raw materials are assessed for their criticality to product safety, efficacy, and supply; just as important in this exercise is mitigation of contamination risk possibly coming from raw materials. The identified risks are related to a) biosafety, e.g., adventitious viruses, mycoplasma, and microorganisms; b) toxins, e.g., endotoxin, bacterial exotoxins, mycotoxins; c) heavy metals; and d) nutritional and metabolic risk. Further, influence of raw materials on cell culture process performance is not always known, making it difficult to establish process-relevant critical raw material attributes as well as specifications beyond those stipulated by certain pharmacopoeia. One of the highest risks is coming back to supply and single-source materials.
Applicable Technologies for Mitigation of Contamination Risk
In order to mitigate risk of a potential contamination during processing, efforts are ongoing in the field of raw materials and suppliers and in introducing media treatment technologies. Some of these activities are briefly outlined below.
Gamma Irradiation
Gamma irradiation is a very common treatment for bovine serum used in biologics and in vaccine production, and viral inactivation efficacy has been demonstrated with this method. However, efficiency on small naked viruses requires a high irradiation dosage [6]. As indicated in Table 2, implementation of such a process is quite lengthy.
Table 2. Pathway towards Implementation of Irradiated Serum in Commercial Processes
Throughout the evaluation process, it has been demonstrated that, although there is no impact on overall process performance, productivity, and product quality, there is a potential that cells may grow slowly in the presence of irradiated serum as presented in Figure 1. Data generated for all model types are comparable and growth delay has been confirmed at scale.
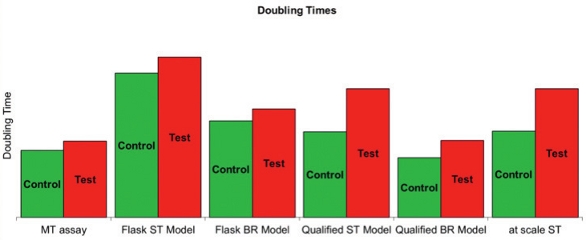 Figure 1. Impact of irradiated serum on cell growth during the seed train process using different models: microtiter (MT) assay, flask seed train (ST) model, qualified ST model, qualified bioreactor (Br) model, and at manufacturing scale ST.
Figure 1. Impact of irradiated serum on cell growth during the seed train process using different models: microtiter (MT) assay, flask seed train (ST) model, qualified ST model, qualified bioreactor (Br) model, and at manufacturing scale ST.Short Wavelength Ultraviolet-type C Light Media Treatment
A short wavelength ultraviolet light (UVC) irradiation remains an attractive option to treat media for mammalian cell culture processes to inactivate any potential viruses and to substantially reduce risks of viral contamination. Ultraviolet lamps produce UVC or “germicidal UV” radiation that is concentrated in the 254-nanometer (nm) region to take full advantage of the germicidal properties of this wavelength. UVC treatment is commonly used in the water treatment industry, but for the biotech and vaccine industries, UVC media inactivation can be particularly attractive, since it is non-intrusive and can potentially handle high-throughput requirements of production facilities. The destruction of viruses using UVC involves a number of pathways, all of which lead to some form of viral genome degradation and/or alteration.
One of the most common mechanisms of degradation involves the formation of a cyclobutane pyrimidine dimer (CPD) within the nucleic acid sequence [7,8]. Since viruses lack the repair enzymes, the genetic damage remains irreversible and, as a result, the virus is inactivated. UVC irradiation has been shown to be effective for a wide range of virus types, including both single-stranded and double-stranded DNA viruses, as well as enveloped and non-enveloped viruses [9,10]. In-house evaluation of this technology has shown it to be particularly efficient in inactivating small, non-enveloped viruses and difficult-to-inactivate viral contaminants (data not shown), making it complementary to gamma irradiation discussed above.
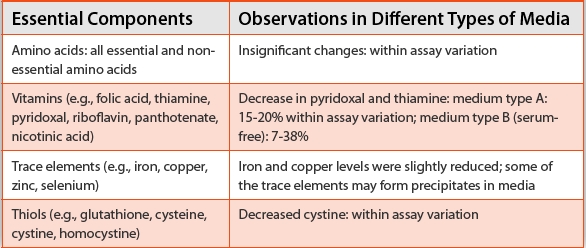
The effect of the use of UVC irradiated media on the cell growth and product quality profile cannot be predicted due to the fact that ultraviolet treatment can possibly cause degradation of small molecules [11] or proteins [12,13], or can generate ion radicals that can be detrimental to mammalian systems. Implementation of UVC-treated media should be preceded by investigative studies to detect any degradation of media components after UVC treatment and impact to the process itself. Based on results obtained during analytical studies as indicated in Table 3, degradation of media components during UVC treatment is marginal for both serum and serum-free media with some exceptions such as thiamine and pyridoxal, which contain heterocyclic structures amenable to photochemical reactions.
Table 3. Tabulated Conclusion Obtained from UvC-treated Media Analytics Studies (within UvC dose range 200-1200 J/m2, for both serum and serum-free media)
Following media analytics studies, cell-based studies were initiated using different small-scale models to evaluate impact on process and productivity. Generated data suggest that use of the UVC-treated media in the cell culture process has no impact on either upstream and downstream process performances or product quality. Additional viral inactivation studies (i.e., spiking studies) are ongoing to validate the effectiveness of viral inactivation using actual model settings.
Nanofiltration of media
Nanofiltration is a robust technique for virus clearance that is commonly applied during downstream biotechnology processes for the purpose of removing potential contaminating viruses. Virus removal by nanofiltration is achieved by a size exclusion mechanism where nanofilters of 20-nm pore size or less effectively remove even small and/ or difficult to inactivate viral contaminants, such as parvoviruses (e.g., minute virus of mice [MVM]), whereas some smaller viruses, such as circoviruses, could potentially pass through the 20-nm nanofilters.
The main limitations of nanofiltration for media application are the consumable cost and the relative slow flux. In addition to process limitations, some large-size media components may potentially be filtered out.
In order to evaluate this technology, a small-scale performance study was conducted using two different nanofiltered serum-free media involved in the process. The selected nanofiltration parameters were then implemented at scale for the production of live attenuated virus (vaccine process). The results obtained indicate that the nanofiltration has no impact on cell growth, viability, or productivity, as indicated by the example data set presented in Figures 2-5. Further, biochemical analytical data indicated that there is only a marginal decrease in concentration of medium components.
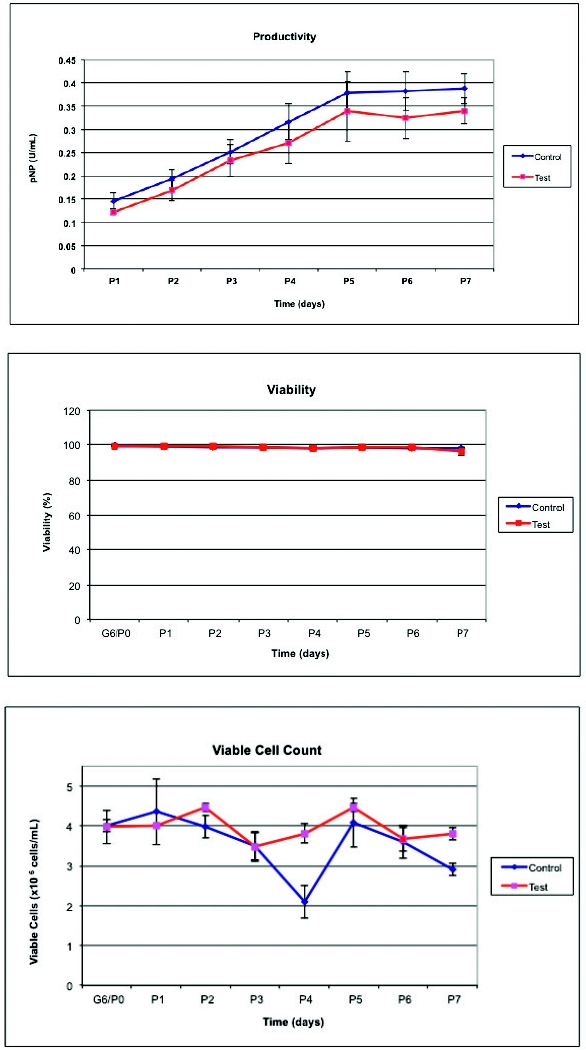 Figure 2. Graphical presentation of generated data using nanofiltered media in small-scale cell perfusion-based model indicating viable cell count, viability, and productivity when compared to control, nonfiltered media.
Figure 2. Graphical presentation of generated data using nanofiltered media in small-scale cell perfusion-based model indicating viable cell count, viability, and productivity when compared to control, nonfiltered media.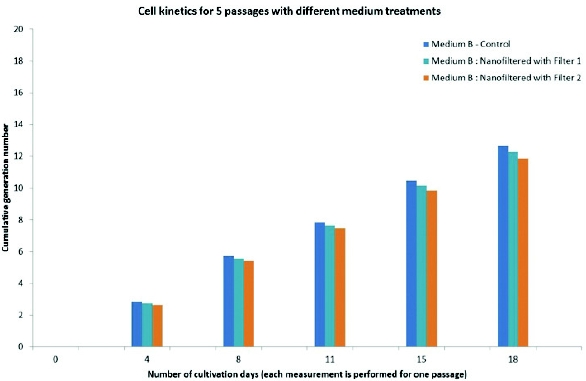 Figure 3. Graphical presentation of cell growth kinetics for 5 passages comparing nanofiltered and control serum-free medium at small scale.
Figure 3. Graphical presentation of cell growth kinetics for 5 passages comparing nanofiltered and control serum-free medium at small scale.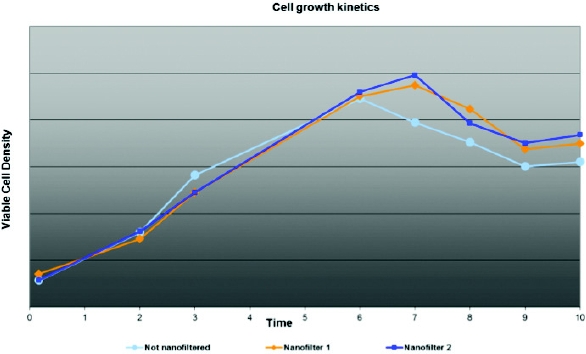 Figure 4. Graphical presentation of cell growth kinetics comparing 2 nanofilters and control serum-free medium at small scale.
Figure 4. Graphical presentation of cell growth kinetics comparing 2 nanofilters and control serum-free medium at small scale.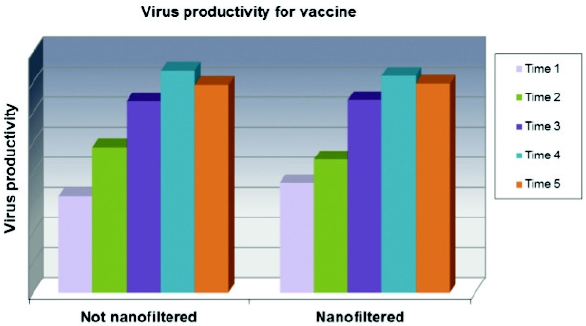 Figure 5. Virus production comparing production using nanofiltered and control serum-free medium at scale.
Figure 5. Virus production comparing production using nanofiltered and control serum-free medium at scale.Replacement of Animal Origin Component by Recombinant Form
Replacement of animal origin components by non-animal forms presents an attractive alternative to address biosafety concerns. However, a major consideration is how non-animal origin raw materials are qualified for use in production of biologics. In that scope, replacement of porcine trypsin by recombinant form would fit in this paradigm. Trypsin is a widely used serine protease for cell detachment, typically isolated from porcine pancreas, and it specifically cleaves at the C-terminus of arginine and lysine within a peptide chain. A gene has been chemically synthesized for the amino acid sequence of the porcine trypsin and transformed into the expression host, which further expressed the recombinant trypsin as active protease with identical properties as the native one. These properties of recombinant trypsin have been confirmed throughout extensive cell culture studies, leading to successful implementation into the commercial process.
A Holistic Viral Risk Mitigation Approach
The holistic risk mitigation approach, as presented in Figure 6, is based on selection and implementation of best practices across the whole manufacturing process ensuring viral safety. This strategy includes extensive virus seeds (in the case of viral vaccines) and cell banks characterization and testing; viral clearance evaluation for downstream processes; extensive control of raw materials and rigorous in-process testing and controls; and sustained vigilance by developing control strategies around raw materials, removal of primary animal-derived raw materials, assessment of risk from secondary animal-derived raw materials, building robust viral barriers around the process, implementation of fast and sensitive in-process assays, and understanding of clearance mechanisms. These actions resulted in a proven safety as there were no further reported cases of an infectious agent transmission. However, most of these actions are challenging to implement because they require extensive analysis throughout small and at large scale studies due to possible impact on process performance and product quality attributes. Furthermore, the need for process validation and regulatory impact must be carefully assessed prior to study execution.
 Figure 6. Holistic contamination risk mitigation approach: Consideration of best practices.
Figure 6. Holistic contamination risk mitigation approach: Consideration of best practices.Conclusion
In order to minimize the risk of viral contamination associated with raw materials, it is suggested to take a holistic approach, i.e., a) developing control strategies around raw materials; b) performing a comprehensive and detailed vendor qualification; c) understanding the impact of selected treatment technology on raw material and further impact to process; and d) developing control strategies around processes to understand the impact on process performance and product quality by defining and executing a comprehensive biosafety strategy resulting in robust viral barriers around the process and in the implementation of fast and sensitive in-process assays. Therefore, UVC media treatment and nanofiltration of media prior to use, combined with a thorough risk assessment and management, are orthogonal approaches that bring pivotal value in leveraging viral safety for biotechnology processes.
References
- R.L. Garnick. Experience with viral contamination in cell culture, in Viral Safety and Evaluation of Viral Clearance from Biopharmaceutical Products, F. Brown and A. Lubiniecki, Editors. 1996. pp. 49-56.
- R.L. Garnick. Raw materials as a source of contamination in large-scale cell culture, in Safety of Biological Products Prepared from Mammalian Cell Culture, F. Brown, et al., Editors. 1998. pp. 21-29.
- J. Skrine. A biotech production facility contamination case study--minute mouse virus. PDA J. of Pharm. Sci. and Technol./PDA 2011; 65(6): 599-611.
- M. Moody, et al. Mouse Minute Virus (MMV) Contamination—a case study: detection, root cause determination, and corrective actions. PDA J. of Pharm. Sci. and Technol./PDA 2011; 65(6): 580-588.
- J. Chen; J. Bergenvin; R. Kiss; G. Walker; T. Battistoni; P. Lufburrow; H. Lam; and A. Vinther. Case study—a novel bacterial contamination in cell culture production—Leptospira licerasiae. PDA J. Pharm. Sci. Technol. Nov-Dec 2012.
- R.W. Nims; G. Gauvin; and M. Plavsic. Gamma irradiation of animal sera for inactivation of viruses and mollicutes—a review. Biologicals 2011; 39(6): 370-377.
- D.L. Mitchell. The relative cytotoxicity of (6-4). Photoproducts and cyclobutane dimers in mammalian cells. Photochem. Photobiol. 1988; 46: 51-57.
- C.J. Bennett; M. Webb; D.O. Willer; and D.H. Evans. Genetic and phylogenetic characterization of the type II cyclobutane pyrimidine dimer photolyases encoded by Leporipoxviruses. Virology 2003; 315: 10-19.
- N.R. Kallenbach; P.A. Cornelius; D. Negus; D. Montgomerie; and S. Englander. Inactivation of viruses by ultraviolet light. Curr. Stud. Hematol. Blood Transfus. 1989; 56: 70-82.
- H. Hermann; R. Buriana; and R. Waldmann. Virus inactivation in foetal calf serum by a combined treatment of gamma-irradiation and UVC irradiation. Animal Cell Technology Meets Genomics 2005. pp. 555-559.
- A.S. Ansari; S. Tahib; and R. Ali. Degradation of phenylalanine in the presence of hydrogen peroxide. Cellular and Molecular Life Sciences 1976; 32: 573-574.
- C. Lorenz; B.M. Wolk; C. Quan; E.W. Alcala; M. Eng; D.J. McDonald; and T.C Matthews. The effect of low intensity ultraviolet-C light on monoclonal antibodies. Biotech. Progress. 2009; 25: 476-482.
- H. Chan; P.R. Gaffney; M.D. Waterfield; H. Anderle; H.P. Matthiessen; H. Schwarz; P.L. Turecek; and J.F. Timms. Proteomic analysis of UVC irradiation-induced damage of plasma proteins: serum amyloid P component as a major target of photolysis. FEBS Letters 2006; 580: 3229-3236.
Author Biographies
Sofie Goetschalckx graduated with a degree in Biochemistry Sciences from the University of Antwerp, Belgium, in 2000. Her professional career started at Pharming N.V., where she was a member of the R&D organization. Shortly after Genzyme’s acquisition, Sofie became responsible for the startup of the cell culture team and establishing new laboratories. She played a pivotal role in the technical transfer, scale-up, and validation of the cell culture processes for Myozyme and Campath. Her team has developed and qualified small-scale models for various large-scale cell culture processes and is still providing global support to Genzyme’s Manufacturing Sites. The important part of Sofie’s work is exploring innovative tools and processes and evaluation towards at scale manufacturing processes.
Virginie Fabre joined Sanofi Pasteur in 2005, where she is currently Unit Head of Downstream and Formulation Processing within Bioprocess R&D. She is in charge of the development of drug substance and drug product processes for vaccine antigens (i.e., recombinant proteins expressed in bacterial hosts and viral vaccines produced in mammalian cell cultures). Virginie has strong experience in virus purification and inactivation. She holds a MSc of Biochemistry and Biotechnology from the National Institute of Applied Science (INSA) in Lyon.
Marijke Wynants is a Process Engineer and a member of the Cell Culture Manufacturing Science team at Genzyme Geel. Prior to joining Genzyme, she was a Ph.D. researcher at KULeuven University in Belgium. She received her Ph.D. in Biomedical Sciences working with different types of cell lines and primary cells.
Landry Bertaux joined Sanofi Pasteur in 2005, where he is currently Scientist in Cellular and Viral Culture unit within Bioprocess R&D. He is in charge of the development of upstream processes for vaccine antigens (i.e., virus and recombinant proteins produced in mammalian cell cultures). He holds a MSc from the Superior High School of Biotechnology in Strasbourg (ESBS) and a MPhil in Molecular and Cellular Biology from Louis Pasteur University in Strasbourg.
Mark Plavsic is a Head of Product Biosafety at Genzyme responsible for setting strategic goals and standards for viral and microbiological safety of biological products and associated manufacturing processes. Previously, Mark was in charge of gene therapy development at Genzyme, overseeing the upstream, downstream, and analytical developments of viral vectors used in gene therapy applications including the process and assay transfer to a GMP production facility. Prior to Genzyme, Mark held senior positions with AstraZeneca, Q-One Biotech, and Life Technologies Corporation. Mark’s education includes doctor of veterinary medicine (DVM) degree, MVSc degree in virology and immunology, and Ph.D. degree in virology and immunology. Mark is a board certified veterinary virologist with the American College of Veterinary Microbiologists (ACVM).
Dr. Otmane Boussif is currently Head of Downstream and Formulation Processing, Bioprocess R&D. His main missions are the development of suitable vaccine purification and formulation processes from Preclinics up to Technology Transfer to manufacturing sites. His main areas of expertise are vaccines and biologics (MAbs, ADCs, recombinant proteins, viruses, gene therapy products). Otmane has more than 20 years of biotech experience spanning from nucleic acids delivery, MAbs purification, and formulation to adjuvanted combo vaccines. Among the companies he worked for are Transgene, Merck-Serono (former Serono), Aventis, Sanofi, and Sanofi Pasteur. Throughout his career, he has held several positions with increasing scientific and management responsibilities. Otmane holds a Master’s degree in Organic Chemistry and Supramolecular Chemistry and Ph.D. in biophysico-chemistry from Louis Pasteur University, Strasbourg.
Dr. Lada Laenen graduated with a degree in Biotechnology Sciences from the University of Zagreb, Croatia, in 1998 and obtained her doctoral degree in Natural Sciences in 2001 at the University of Kaiserslautern, Germany. Lada has more than 15 years’ experience in the biopharmaceutical industry. Her professional career started at PLIVA Inc., Zagreb, Croatia, and continued at MIXIS Genetics, Paris, France, in different development roles focusing her work on antibiotic production. After her move to Belgium, she established the research department at Flen Pharma NV with focus on tissue cultures and clinical studies in collaboration with different Belgium hospitals, and then moved to the Johnson & Johnson Tibotec/Virco branch, where she worked in the HIV field. In 2007, she joined Genzyme, and following the Sanofi acquisition in 2011, she headed the Cell Culture and Microbiology departments, focusing on innovation, process development and validation, regulatory submissions, and providing support towards commercial manufacturing.