This is the third article in a series of publications titled “How Clean is Clean in Drug Manufacturing?” The first and the second articles introduced the term Equipment Cleaning Process (ECP); examined residue limits setting, including health based considerations; and, showed applicability to Cleaning Validation Practices of the Threestage Process Validation Concepts described in the FDA Process Validation Guidance. This article will continue the discussion about a risk based and lifecycle approach to Cleaning Validation. We will discuss assessment of variation in ECP and tools for benchmarking the culture of Cleaning Validation at drug manufacturing facilities. In a recent article, Dr. Mike Long asked a simple question: “How much variation is acceptable in our products and processes?” Although the question is simple, answering it could be very complex. It requires understanding of product, process, and those factors that impact variability. Very early on in the lifecycle of any process, including ECP, risk tools should be used to help understand and control the amount of variation.
Line of Sight Approach
Ensuring process robustness through the full lifecycle requires that we intentionally determine the maximum amount of process variation we are willing to accept. As Dr. Long recommended in the article cited above, a “Line of Sight” approach could be applied to achieve this level of process understanding. The amount of testing we perform during Stage 2 (Process Performance Qualification) and Stage 3 (Continued Process Verification) must have a clear path back to the attributes of the product-process we endeavor to deliver. It is also recommended to have a risk-based and statistical justification for a meaningful analysis. The following steps, which embedded within Stages 1 (Process Design and Development) and 2 (PPQ), provide the basis for this “Line of Sight” approach (Figure 1).
 Figure 1. Line of Sight: CQA Identification to PPQ Sample Size Selection.
Figure 1. Line of Sight: CQA Identification to PPQ Sample Size Selection.Perform a Risk Assessment
Some of the same principles and practices typically applied to a manufacturing process can be readily applied to cleaning validation. First, one must identify Critical Quality Attributes (CQAs). Typical CQAs for residue limits are listed in Figure 2. These are types of residues for which measurable levels must be determined that are to be safe, acceptable and achievable on equipment surfaces used in the manufacturing of drug substances and drug product and being cleaned by ECP. Residue types include chemical, physical, and biological attributes.
 Figure 2. Cleaning Validation Residuals Critical Quality Attributes.
Figure 2. Cleaning Validation Residuals Critical Quality Attributes.Another factor of Cleaning Validation CQAs includes surfaces being cleaned, such as:
- product contact surfaces; and,
- surfaces that are in close proximity to the product, such as autoclaves and lyophilizers.
- surfaces that make up manufacturing facilities (its floors, walls, and ceilings) may be considered, but treated according to their risk level to possible cross-contamination. Typically, visually clean criterion should suffice for most of these surfaces in the facilities, other than highly active compounds.
A risk based approach should be based on proximity and/or possibility of cross-contamination to the product. All of these surfaces are subjects of Cleaning Validation studies. After the initial CQA assessment, the relative risk of the individual CQAs needs to be determined. Not all Quality Attributes are equivalent from a risk standpoint. The impact of the failure of a particular CQA has a place in a continuum of criticality. Some CQAs will have a lower impact on patient safety compared to other CQAs along that continuum that have a more significant impact.
For example, the criticality and risk to patient safety of particles and bioburden in a sterile, parenteral drug formulation is much greater than the same particles and bioburden in a non-sterile oral formulation. The relative difference needs to be assessed with a method called the “Continuum of Criticality,” which will ultimately provide the basis for the extent of Cleaning Validation studies and PPQ sample size justification. The continuum of criticality assessment is fairly simple from a conceptual standpoint. CQAs that have a high severity rating should have a higher standard of testing applied to them as compared to those CQAs that have been determined to have a lower severity. An example of such a criticality assessment based on keywords is shown in Table 1.
Table 1. Example of Quality Attributes Criticality Assessment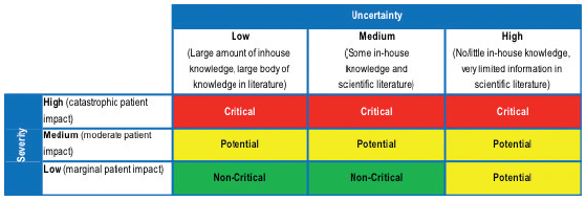
Table 2 illustrates how relative criticality assessment is performed for surfaces being cleaned where those surfaces that have potential for immediate product contamination would require more extensive Cleaning Validation studies (including number of samples taken) than those that are in close proximity to the product or those that constitute structure of the facility.
Table 2. Example of Surface Being Cleaned CQA Continuum of Criticality Analysis with Relative Extent of Validation Studies/ Statistical Sampling Requirements.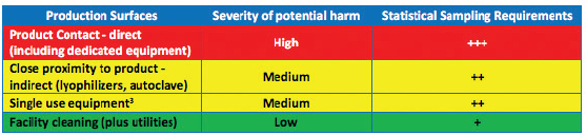
Next, Table 3 shows an example of a rationale for selection of swab locations for a piece of equipment.
Table 3. Example of a Rationale for Selection of Sample Swab Location Using Risk Assessment Tools.
In the following Table 4 is shown an example of application of a use of rationale presented in Table 3 for a selection of swab locations in a vessel.
Table 4. Example of a Rationale for Selection of Sample Swab Location for a Vessel using FMEA-like Risk Assessment.
An important example of a Risk Assessment exercise that Cleaning Validation can benefit from is an examination of Bioburden concerns, such as evaluation of risks that could impact Clean Hold studies. Figure 3 shows an illustration of an example of some of those elements that should be considered when one evaluates Clean Hold risks.
 Figure 3. Example Clean Hold Risk Assessment Points to Consider.
Figure 3. Example Clean Hold Risk Assessment Points to Consider.Another important kind of attribute that should be determined during Cleaning Validation risk assessment is health based residue limits based on API toxicity information. An example of such assessment is depicted in Table 5. It is recommended that more extensive studies with additional sampling be performed on the cleaning processes that are designed to remove API’s with lower Acceptable Daily Exposure values, as they may have a higher hazard potential for the patient.
Table 5. Example API Toxicity CQA Continuum of Criticality Analysis with Relative Extent of Validation Studies/Statistical Sampling Requirements.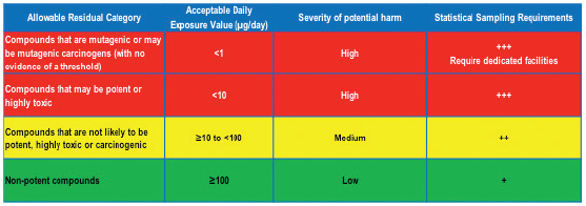
Use of Knowledge Management and Level of Cleaning Validation Maturity
Table 6. Cleaning Validation Program Based on 3-Stage Approach 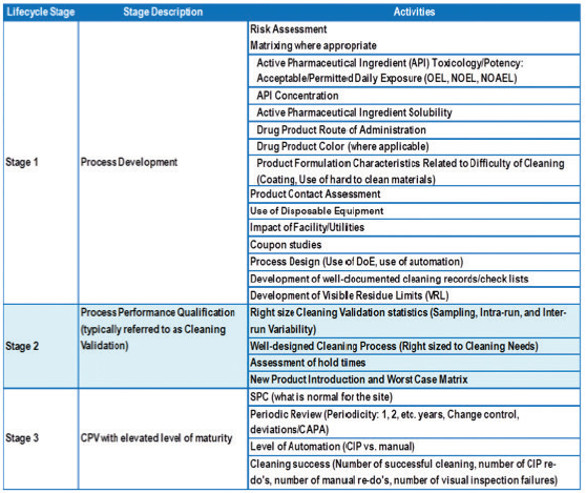
The “Line of Sight” approach allows us to develop Cleaning Validation programs that use not just the risk management, but also knowledge management tools. As outlined in ICH Q10, Knowledge Management is an enabler for successful implementation of pharmaceutical quality systems just as is Quality Risk Management. Therefore, the level of knowledge that we acquire, analyze, leverage, and discriminate prior, during, and after Cleaning Validation studies is directly corresponding to a term we call a “Level of Cleaning Validation Maturity” at the manufacturing facilities. For instance, Table 6 outlines a typical Cleaning Validation program at pharmaceutical or biopharmaceutical sites that is based on an approaches following FDA’s Cleaning Validation Guidance and similar in design to one outlined in FDA’s Guidance for Process Validation. It is evident that recommended activities in Stage 1 are more extensive as we gather data regarding new products and processes being introduced in our production. The more meaningful data (i.e., certainty) we have on new substances and their cleaning, the less risk (i.e., uncertainty) we take during development and implementation of cleaning processes. We extrapolate this level of knowledge onto the entire cleaning process continuum. This correlation of Cleaning Process (ECP)/ Validation knowledge and Level of Cleaning Validation Maturity is shown in Figure 4.
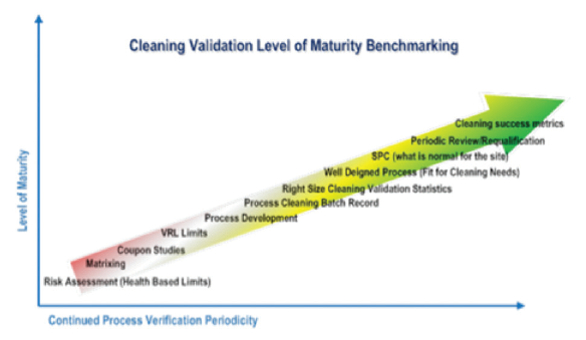 Figure 4. Cleaning Validation Level of Maturity
Figure 4. Cleaning Validation Level of MaturityConclusions
This article illustrates application of risk-based lifecycle approach to Cleaning Validation specifically applying ICH Q9 and Q10 principals. Authors illustrated usage of Quality Risk Management tools to define, measure and assign outcomes of QRM exercises to Stages 1, 2 and 3 of Cleaning Validation. In addition, the article introduces term “Cleaning Validation Level of Maturity” which should help practitioners to benchmark their programs, solidify and improve them if required.
References:
- Mike Long, “Risk and Statistics Serve as Tools for Solving Variation Riddles”, PDA Letter, April, 2013
- PDA Technical Report No. 60: Process Validation: A Lifecycle Approach; Parenteral Drug Association: 2013. www.pda.org/bookstore (accessed March 14, 2013), Bethesda, MD 2013
- Igor Gorsky, How Clean is Clean in Drug Manufacturing?, American Pharmaceutical Review, May/August 2014
- Igor Gorsky, How Clean is Clean in Drug Manufacturing? Part 2, American Pharmaceutical Review, January/February 2015
- FDA Guidance For Industry, Process Validation: Principles and Practices, January 2011