The Purpose of the Compendial Test The USP Antimicrobial Effectiveness Test (AET) is a product quality test which is designed to be, so far as is possible, a reproducible biological measurement of the activity of the preservative system in a product. This test is required for multi-dose presentations of pharmaceuticals as well as anhydrous ointments that contain a preservative system [1].
The purpose of the test drives its experimental design, a design which also imposes some limitations to the application of the results of the AET. The antimicrobial effectiveness test first appeared as a USP General Chapter in the 18th revision, official September 1, 1970. This chapter, from its beginning, was designed to evaluate the performance of antimicrobials added to inhibit the growth of microorganisms that might be introduced during or subsequent to the manufacturing process. Its purpose, like all USP microbiology tests numbered under 1000 (for example <71> Sterility Tests <61>, Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests <62> Microbiological Examination of Nonsterile Products: Tests for Specified Microorganisms Sterility < 85> or Bacterial Endotoxins Test), is to demonstrate compliance with monograph requirements as described in the National Formulary [2, 3]. It is not designed to be a check of the ability of the packages final product to withstand in-use contamination.
 Figure 1. Antimicrobial Effectiveness Test
Figure 1. Antimicrobial Effectiveness TestThe Antimicrobial Effectiveness Test (AET) is a suspension test for microbial kill. In this design a controlled inoculum of the challenge organism(s) is placed in suspension with the sample to be tested, and then the number of survivors determined at different time points (Figure 1). This simple design has benefits in terms of the opportunities to compare the biological activity of a wide array of preservative systems in a controlled manner, but some significant limitations as well. As just one example, the preservation of a finished product is influenced not only by the product’s preservative system but also the patient’s use of the product, an influence heavily affected by the container/closure properties of the packaging. The measurement of this in-use potential for contamination of the finished product is outside the scope of the compendial AET (see below).
There are several variations to the standard AET that have been described in regulatory and trade standards [4-6]. This review will be restricted to the tests commonly used in the pharmaceutical, OTC and medical device industries with emphasis on the USP test [1].
The compendial tests of the USP and the European Pharmacopeia have very similar methods, but they differ in sampling points and acceptance criteria. [6- 8]. In general, both compendia follow a pattern of analyzed product risk – more patent risk for a product category is associated with increased (and shorter) testing intervals over the 28-day period, and more stringent log10 unit reduction requirements for viable CFU from the inocula (for example, see Tables 1 and 3 in reference 1).
Demonstration of method suitability
The measurement of microbial kill requires the ability to measure the number of surviving microorganisms with time after exposure to the antimicrobial agent. However, carryover of residual preservative from the test could inhibit growth in the recovery medium, leading to poor microbial recovery. This potential residual activity must be neutralized and it is necessary to demonstrate the adequacy of neutralization for these tests [9-12]. This demonstration of neutralization in compendial microbiological tests is known as demonstration of method suitability.
As with the overall design of the AET, it is important at the outset to clearly define the goal of the method suitability study (as described in the compendia). This goal is not to demonstrate the ability to recover microorganisms present in the product. If that were the case then the challenge organisms would be inoculated directly into the product – they are not. This direct inoculation of the product will not work if the antimicrobial properties of the product are strong as the product could well kill off all challenge organisms before it was possible to plate the test organisms. The goal of a microbiological method suitability test is to demonstrate that any residual antimicrobial properties of the product or the recovery method have been neutralized, using the challenge microorganisms as a kind of biological indicator of neutralization.
The overall purpose of a neutralizer evaluation (Method Suitability Study) is to serve as a control experiment to the preservative system evaluation (AET). Therefore, replication of the critical parameters of the AET is critical to the neutralizer evaluation (see below for determination of “critical parameters” in the discussion of Laboratory Investigations). A good overall reference for microbial recovery studies can be found in USP < 1227> “Validation of Microbial Recovery from Pharmacopeial Articles” [13].
It is important to note that although at the present time there is not a method suitability test written into the compendial AET chapter, basic GMP requirements call for evidence that a test is suitable for its intended purpose. Specifically, 21 CFR 211.194 Laboratory Records states in section (a)(2) “(If the method employed is in the current revision of the United States Pharmacopeia, …and the referenced method is not modified, a statement indicating the method and reference will suffice [for validation of the assay]). The suitability of all testing methods used shall be verified under actual conditions of use.” In other words – a method suitability test.
Microbiology Laboratory Investigations and Proactive Documentation
Investigations are a critical area of regulatory concern and there exists a great deal of confusion about regulatory expectations in this regard. This confusion is especially noticeable in the area of Microbiology laboratory investigations. The FDA has provided guidance on the topic, drafting the “Guidance for Industry - Investigating Out of Specification (OOS) Test Results for Pharmaceutical Production” [14]. Interestingly, this guidance document only briefly touches upon microbiological data, stating in section IV.C.1.a. that “…In the case of microbiological assays, the U.S. Pharmacopeia (USP) prefers the use of averages because of the innate variability of the biological test system”. While this is interesting and undoubtedly true, it does not help in designing a laboratory investigation. This silence on the part of the guidance document is explained in footnote 3, which explicitly excludes biological studies from the scope of the document (this statement includes microbiological studies).
This guidance document, although written for analytical chemistry tests, does provide useful guidance to microbiology laboratory investigations. For example, the FDA guidance’s focus on investigating a potential laboratory component (with its respective responsibilities) prior to the full scale investigation is especially helpful. Great care should be taken, however, in over-applying these documents into areas for which they are not appropriate.
Table 1. Example SOP Listing to Support Antimicrobial Effectiveness Testing

This leaves us with the need to develop our own laboratory investigation procedures. The initial question in any potential test failure is to determine the validity of the test result. This is especially true of the AET which will be conducted over the period of a full month and may include contributions from several different laboratory technicians. It is therefore critical to realize that the lab investigation for any microbiological test is heavily dependent on proactive documentation acquired during the test. The GMP microbiology lab collection of this documentation is aided by a well-organized SOP system [15]. An example of such a SOP support system that might be useful in an AET laboratory investigation is provided in Table 1.
This well-organized system also allows for easy identification of the necessary proactive documentation for the GMP compliant AET. A useful exercise in this regard is to review the USP chapter as well as your internal AET procedure, making a list of every time either document mentions a microbial growth medium, a piece of equipment, a stock culture or a number (temperature range, duration, frequency, CFU/plate). This list becomes your checklist for “Critical Factors” (if the parameter is not critical, why specify it?). With this list in hand, pick a past AET at random and confirm that all critical items are included in the proactive documentation. Any failures in documentation found by this gap analysis should be addressed immediately to assist in future investigations (and GMP audits!). Extensive documentation of compliance with the laboratory’s SOP system increases the chances of a successful AET investigation.
All investigations should happen in a timely fashion. The potential AET failure should be communicated immediately to the laboratory supervisor. The investigation should then follow an established procedure, a procedure that was developed in advance with the approval of the Quality Assurance Unit (QAU).
The first step of a laboratory investigation is to check for the easy issues. Look for common data entry errors:
- Incorrect math
- Sample dilution series error
- Transcription error
- Clerical error
If these are found, correct the error under your procedure for such corrections. If the lab investigation does not identify correctable data entry errors as the source of the putative AET failure, then a more complete investigation should be initiated (see Figure 2 for an example of a possible model for such an AET laboratory investigation). The overall laboratory investigation should be coordinated though the QAU as per 21 CFR 211.160.
 Figure 2. Flow Chart of Example AET Laboratory Investigation
Figure 2. Flow Chart of Example AET Laboratory InvestigationThe complexity of the AET as a laboratory process (despite its simple design) requires that the investigation examine a variety of different topics as outlined in Figure 2. The evaluation of data records and test samples will play a large part of this process. The value of extensive proactive documentation was described above, but this is not sufficient for a complete AET investigation. It is also of value to ensure that the samples showing growth are in fact the challenge organism (and not a “resistant” contaminant) and that the product samples are correct. Physical constraints on space may prevent retention of all samples and test articles that might be of interest. However, it is prudent to retain as many of the critical samples as possible until the successful completion of the test to aid in the laboratory investigation of a potential issue.
Specific attention should be given to “confirmatory testing”. As noted above, the AET is conducted over the period of a full month and may include the contributions of several technicians. The inherent variability of this test is obvious and it is reasonable to expect a marginally preserved formulation to fail on occasion due to random chance. In the author’s opinion, this specific situation allows for an argument to be made in favor of “confirmatory testing”2. This decision to go forward with “confirmatory testing” should be guided by SOP - assay variability is a known quantity in microbiological testing and procedures should be in place to resolve indeterminate results.
In addition, the biology of the test must be considered in the interpretation of the data from that AET. For example, a product that shows no reduction of Aspergillus brasiliensis for 14 days, and then shows an increase at day 28 is unlikely to be a true failure of the USP criteria. This situation might be the result of spore “clumps” in the inoculum breaking apart, yielding an apparent increase in the CFU. The sample should be examined microscopically for the presence of hyphael growth – spores do not reproduce, and a hyphael mat is unlikely to increase the numbers of colony forming units on a nutrient agar plate.
The final phase in the laboratory investigation is to close it. The investigator should determine if the AET failure was the result of:
- Laboratory error (invalid test)
- Assay variation
- Failing product
- Inconclusive causes
If the AET failure is determined to be due to error in lab practice, then the test may potentially be declared invalid. An invalid test is, by its nature, not a valid test and therefore the subsequent test is not a retest but rather the first correct performance of the assay. If the test is invalid, a valid test should be run immediately to determine compliance.
This determination requires identification of a root cause for the lab error as well as a corrective action plan to be put into place in the laboratory to remedy the error and prevent its recurrence. This plan should include a method to measure the efficacy of the corrective action. It has to be stressed that clear evidence of lab error is required to determine the test invalid. If the investigation is inconclusive, the data must be held to be valid.
If the putative lab failure was due to assay variation, a determination should be made as to the appropriateness of confirmatory testing (as discussed above). In the conclusion of the laboratory investigation there must be a clear rationale of how confirmatory testing (if used) is factored into the final determination of the microbial quality of the preservative system
If the result of the investigation is that the product did indeed fail, then the product should be handled as appropriate under management guidance for OOS.
If the laboratory investigation is inconclusive, the test results are considered valid. It is important to stress the point again that positive evidence is required to invalidate a microbiological laboratory test. All test results are assumed correct unless there is unambiguous evidence of a laboratory error of sufficient impact to invalidate the test.
A general discussion of investigations can be found in USP chapter “< 1117> Best Microbiological Practices” [16]. The USP notes that interpretation of microbiological data is difficult and strongly recommends that the supervisor of the lab should be one with academic training in microbiology or bacteriology. Microbiological data requires interpretation; it is not data that can be presented solely by test results. Investigations should be conducted from a broad perspective, not only looking at equipment, training, product and test, but also the underlying biology associated with the situation.
Other Considerations
There are a variety of associated questions and topics surrounding the determination of antimicrobial effectiveness for a preservative system. Among these are the use of additional challenge organisms to stress different aspects of the preservative system and the determination of antimicrobial efficacy under simulated in-use conditions.
“Extra” Challenge Organisms
Over the years there have been a variety of additional organisms suggested for inclusion in the AET [6]. Recently this effort has gained a great deal of emphasis with the realization that Bulkholderia cepacia is a real health threat to a portion of the population when inhaled, (the susceptible population consists primarily of cystic fibrosis patients and those at risk for pneumonia) [17, 18]. In addition, this microorganism seems adept at overcoming preservative systems and so seems a good choice for a biological challenge [19, 20]. However, the choice of additional challenge organisms beyond those listed in USP is a difficult one – as an example of this complexity the reader is referred to a recent discussion of the wide-spread need for inclusion of B. cepacia in QC testing found in the communications of Torbeck et al [21] and Sutton [22].
If the decision is made to include additional challenge organism(s) in the AET, the additional organisms should be chosen with care. Good potential candidates for in-house AET challenges might be selected by:
- Regulatory concerns (has your facility been asked to do something by an inspector?)
- Product concerns (has an organism caused a failure of the preservative system in one of your products?)
- Facility concerns (has your environmental monitoring or water monitoring programs isolated a specific organism in high numbers or frequency?)
Note that there is no regulatory requirement to add additional microorganisms to the AET. However, if you decide to do so, it is prudent to have a solid, written justification for the organism chosen and a well-defined response if that “additional” AET organism should fail to meet the criteria established for passing the test.
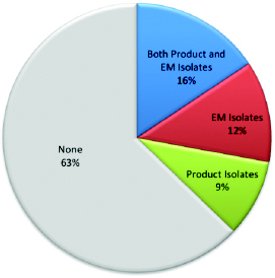 Figure 3. Sources of Additional Microorganisms for AET
Figure 3. Sources of Additional Microorganisms for AETA recent survey benchmarking the practices of companies performing AET addressed this issue3. The survey received 172 complete responses. Of these responses, 108 did not add additional organisms; however, this also means that approximately 37% of the respondents were using additional organisms in the AET. These respondents cited environment monitoring isolates and isolates from products as the sources of the additional organisms (see Figure 3).
Simulated in-use Designs
We have examined the AET as a laboratory test that has value as a QC test but may not be appropriate for determining the behavior of the finished product preservative system in the field. There have been several recommendations for methods to determine the ability of finished product to withstand in-use conditions.
An example of this approach was taken by the cosmetics industry in developing an alternative method from the USP antimicrobial effectiveness test. This assay was then tested by “simulated use” against several other predictive methods, and compared favorably to several types of antimicrobial effectiveness testing methods available at the time [23].
A second example is the EMEA recommendation for in-use testing of preserved products [24]. This topic was reviewed in response to a preliminary draft of the guidance document prior to its finalization [25]. In the authors’ opinion, in-use testing for chemical attributes makes sense if oxidation or evaporation of the preservative is a concern, especially in gas-permeable containers. The effects of repeated microbiological challenges is a reasonable consideration, and should be addressed in the lab by repeated low-level challenge studies, or in the field by simulated in-use studies.
The laboratory-based option using low-level challenges to simulate repeated microbial challenges has been described by Urban et al ([26] who used a battery of challenge organisms with multiple, lowlevel inoculations to model a repeated contamination events in a simulated in-use test. By this method, the sample is challenged repeatedly over the 28-day in-use shelf-life with a low level (102 – 103 CFU/mL) of each organism, and the efficacy of the preservative system is evaluated under this simulated abuse situation. This approach has the advantages of simulating consumer abuse of the product under a controlled laboratory setting and may be designed to monitor the entire in-use shelf life. If this design is used, the volume of product sample used should be large enough to withstand potential dilution of the preservative system by multiple inocula (pooling of sample may be required).
The manufacturer would be well-advised to conduct a simulated inuse preservative efficacy study as part of the product development, but this will have to be done using in-house resources as there are no useful regulatory guidance documents for pharmaceutical products in this area.
Effect of Container/Closure systems
Packaging components, especially dispensing closures, can be important considerations in preventing microbial contamination during consumer use. A study by Brannan and Dille [27] showed that, during consumer use, unpreserved shampoo with a flip-cap had the greatest degree of protection from contamination (0%). for an unpreserved skin lotion, a pump-top dispenser afforded the best protection from contamination (10%). Other types of closures tested included the standard screw-cap and slit-cap. The screw-cap closure provided the least amount of protection, while the slit-cap provided moderate protection from contamination. This study underscores the need for considering preservation as an attribute of the entire product, not just the active preservative.
Conclusions
The Antimicrobial Effectiveness Test is a rather simple suspension test in design. This suspension test design requires demonstration of method suitability to document the adequacy of the microbial recovery system in the presence of residual product. Conducting the AET in a GMP environment requires tight control over the laboratory systems and extensive proactive documentation. This documentation bears additional benefits during an investigation, especially as this test runs over a month-long period and frequently the major material for the investigation consists of the assay documentation. The test may be supplemented by the use of additional challenge organisms chosen carefully for their value, and by additional tests designed to simulate in-use microbial challenges. Finally, a complete picture of the antimicrobial properties of a finished dosage form cannot be determined without consideration of the container closure system and its contribution to the ease of product contamination in-use.
Author Biography
Scott Sutton, Ph.D., is the Principal of Microbiology Network, Inc. (http://microbiologynetwork.com), a company he started in 1996 as a means to encourage training and communications within the microbiological community. He is a recognized consultant and trainer with emphasis in GMP, investigations, Environmental Monitoring and contamination control (both Aseptic manufacturing and non-sterile production facilities) as well as microbiology laboratory audits and operations. The Microbiology Network supplies consulting, training, webinars and e-mail discussion groups. Dr. Sutton is an active author and speaker for the industry, supports PDA and has served with the USP Analytical Microbiology Committee of Experts since 1993. The opinions expressed in this article are his alone and do not necessarily reflect the policies or positions of any organization with which he is associated. He may be reached at [email protected]
References
- USP. 2013.<51> Antimicrobial Effectiveness Test USP 36/NF 31 Vol 1 The United States Pharmacopeial Convention, Inc. Rockville, MD. p. 54-55 official May 1, 2013
- USP. 2013. General Notices and Requirements Section 2.10 “Official Text” USP 36/NF 31 Vol 1 The United States Pharmacopeial Convention, Inc. Rockville, MD. p. 3 official May 1, 2013
- Sutton, S. and R. Tirumalai. 2011. Activities of the USP Microbiology and Sterility Assurance Expert Committee During the 2005–2010 Revision Cycle. Amer Pharm Rev 14(4):12-30.
- Siegert, W. 2012. ISO 11930 - A Comparison to Other Methods to Evaluate the Efficacy of Antimicrobial Preservation. SOFW-J 138(7):43-53.
- CTFA. 2007. CTFA Microbiology Guidelines • M-3 A Method for Preservation Testing of Water-Miscible Personal Care Products • M-4 Method for Preservation Testing of Eye Area Cosmetics • M-5 Methods for Preservation Testing of Nonwoven Substrate Personal Care Products
- Sutton, SVW and D Porter. 2002. Development of the Antimicrobial Effectiveness Test as USP Chapter< 51> PDA J Pharm Sci Tech. 56(6):300-311.
- Moser, C. and B. Meyer. 2011. Comparison of Compendial Antimicrobial Effectiveness Tests: A Review. AAPS Pharm Sci Tech. 12(1):222-226.
- Matthews, BR. 2003 Preservation and Preservative Efficacy Testing: European Perspectives. Eur J Parent Pharm Sci. 8(4):99-107.
- Sutton, S, et al. 2002. Validation of microbial recovery from disinfectants. PDA J Pharm Sci Tech. 56(5):255-266.
- Russell, AD et al 1979. Microbiological applications of the inactivation of antibiotics and other antimicrobial agents. J. Appl. Bacteriol. 46:207-245
- MacKinnon, IH. 1974. The use of inactivators in the evaluation of disinfectants, J. Hyg. (Camb.) 73:189-195.
- Sutton, S. 1996. Neutralizer evaluations as control experiments for antimicrobial efficacy tests IN: Handbook of Disinfectants & Antiseptics. J Ascenzi (ed). Marcel Dekker Publ. pp. 43-62.
- USP. 2013 < 1227> Validation of Microbial Recovery from Compendial Articles. USP 36/ NF 31 vol 1 The United States Pharmacopeial Convention, Inc. Rockville, MD. p. 989-991 official May 1, 2013
- FDA. 2006. Guidance for Industry: Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production.
- Sutton, S. 2010. The Importance of a Strong SOP System in the QC Microbiology Lab J GXP Compliance 14(2):44-52 2010
- USP. 2013 < 1117> Microbiological Best Laboratory Practices USP 36/NF 31 Vol 1 The United States Pharmacopeial Convention, Inc. Rockville, MD. p. 794-799 official May 1, 2013
- Kutty, P., et al. 2007. Multistate Outbreak of Burkholderia cenocepacia Colonization and Infection Associated with the Use of Intrinsically Contaminated Alcohol-Free Mouthwash. Chest 132:1825 – 1831.
- Kuhn, RJ et al. 1982. Bacterial Contamination of Aerosol Solutions Used to Treat Cystic Fibrosis. Amer J Hosp Pharm. 39:308-309.
- Decicco, B., et al. 1982. Factors Affecting Survival of Pseudomonas cepacia in Decongestant Nasal Sprays Containing Thimerosal as Preservative. J Pharm Sci. 71(11):1231 – 1234.
- Borovian, G. 1983. Pseudomonas cepacia: growth in and adaptability to increased preservative concentrations. J. Soc. Cosmet. Chem. 34(4):197-203.
- Torbeck, L., et al. 2011. Burkholderia cepacia: This Decision is Overdue. PDA J Pharm Sci Tech 65(5):535-543.
- Sutton, SVW. 2012. Letter to the Editor in Response to Friedman et al., Burkholderia cepacia: This Decision Is Overdue. PDA J Pharm Sci Tech 66(2):91-95.
- Farrington, J.K., et al. Ability of Lab Methods to Predict In-Use Efficacy Antimicrobial Preservatives In an Experimental Cosmetic. Appl Envir Microbiol. 60(2):4553-4558. 1994.
- EMEA. 2001. Note for Guidance on In-Use Stability Testing of Human Medicinal Products
- Sutton, SVW et al. 1998. In-Use Stability Testing: What Data Are Required and When?. Reg Affairs J 9(10):728-733.
- Urban, S., W. Hecker, and I. Schiller (1981) Zbl. Bakt. Hyg. I. Abt. Orig. B. 172:478
- Brannan, D.K. and J.C. Dille. 1990. “Type of Closure Prevents Microbial Contamination of Cosmetics During Consumer Use.” Appl Environ Microbiol. 56(5):1476-1479.