The use of purified lipopolysaccharide (LPS) as a surrogate contaminant for natural endotoxin currently remains a controversial and misunderstood subject. Soon after the FDA published a questions and answers document1 stating that firms should establish the stability of assayable endotoxins in their products, mixed interpretations of how to achieve this requirement occurred. This was especially true for products containing no measurable endotoxin. There had also been a misunderstanding of the original intent of the FDA requirement. The original intent was clarified at the Bacterial Endotoxins Summit for Low Endotoxin Recovery on September 14-15, 2015 in Iselin, New Jersey. The original intent of the FDA statement was to assure that if a firm’s product had assayable endotoxin, the activity levels would not increase and/or exceed the acceptance criteria at the end of the product shelf life. Some firms interpreted the FDA answer to Question #3 to artificially contaminate a clean product with endotoxin surrogates to comply with the expectation for the demonstration of the stability of assayable endotoxin.
In 2012, Genentech scientists2 were complying with their interpretation of the FDA requirement to demonstrate the stability of assayable endotoxins. As most monoclonal antibodies performed in cGMP environments are highly controlled, the product contained no endogenous endotoxin. They reported that when endotoxin (5 EU/mL) was placed directly into a biological product that contained divalent cation chelating buffers plus polysorbate, the measurable endotoxin activity rapidly disappeared to 2 EU/mL. Therefore, the firm reported that the product matrix did not meet the acceptance criteria for stability of assayable endotoxins. The terms Low Endotoxin Recovery (LER) and endotoxin “masking” were used to describe this phenomenon. The firm also reported that these same test articles, when injected into rabbits, caused a temperature increase in one of three rabbits. They reported that the rabbit pyrogen test failed while the LAL test failed to detect at least 50% of the endotoxin.
Upon further review of the experimental data, the Genentech experiments had been performed using Control Standard Endotoxin (CSE) as a surrogate contaminant. It seemed logical to assume that both Reference Standard Endotoxin (RSE) and CSE are endotoxin as they are the standards used to prepare standard curves and sensitivity titrations endotoxin activity measurements. CSE and RSE are not natural endotoxin that would occur as contamination in products. Natural endotoxin exists as pieces of Gram negative bacteria which contain all of the other cellular wall debris. Natural endotoxins do not exist as purified LPS. Endotoxins may exist as vesicles, blebs, bilayers and other complex cell wall debris. Since only one part of the endotoxin molecule is the biologically active fever producing part of the molecule, the degree of aggregation of the molecules also determines the degree of pyrogenicity both in vivo and in vitro.
To understand the LER phenomenon, which we now understand better as Low Lipopolysaccharide Recovery (LLR) along with its correlation to the rabbit pyrogen test (RPT), one has to examine the implementation of the LAL test as a replacement for the RPT forty years ago.
The discovery and development of the Limulus Amebocyte Lysate (LAL) test by Dr. Jack Levin was enabled using an LPS derived from E. coli3. LPS that was derived from E. coli and Klebsiella sp. were used to compare the relative endotoxin sensitivity of the LAL and RPT tests thus demonstrating LAL’s potential as a screening test for endotoxin pyrogen detection in parenteral products4. As the LAL test was implemented and improved, largely replacing the RPT, there was a naïve assumption that the purified LPS standards that we know as primary RSE and secondary CSE LPS standards behave equivalently to natural occurring endotoxin (NOE) in all product matrices. This assumption is definitely not the case.
The RPT, LAL, and Mononocyte Activation Test (MAT) all measure activity of the endotoxin molecule. The experience of the LAL industry is that the lyophilized LPS standards are reliable sources of standard activity materials, but are more unstable than native endotoxins. RSE is given a shelf life of no more than 14 days and CSE no more than 28 days when reconstituted and stored at stock concentrations at 2-8°C. Most LAL scientists discard diluted standards after an eight hour period of time. The industry is demonstrating that pyrogen free water controls inoculated with CSEs are demonstrating greater stability than 30 days. However, when exposed to product matrices other than pyrogen free water, the LPS molecules behave differently.
Bowers and Tran5 prepared NOE from a variety of Gram negative bacterial sources to assess the stability of unpurified endotoxin across various matrices, temperatures, and containers. It was demonstrated that high EU/ml level solutions of native endotoxins are very stable for long periods of time. These native endotoxins are very useful for assessing endotoxins stability in many product matrices.
It is important to know and understand how the E. coli endotoxin LPS standards were prepared, chosen, and correlated to the RPT. A review of the creation of the RSE and CSE test standards versus the RPT is useful in understanding the extremely variable and less sensitive (1000X) RPT. It is also important to understand the wide variety of endotoxin activities from different Gram negative bacterial sources. A team of researchers at the Max Planck Institute, led by Westphal and Lüderitz, described methods of extracting LPS from a variety of Gram negative bacterial sources6. The creation of the endotoxin standard was led by the FDA and USP. They commissioned Rudbach7 to prepare reference material LPS that was free from proteins, had an approximate average endotoxin activity, and was characterized chemically. This process proved to be quite complicated. In order to retain endotoxin activity, the LPS molecules had to have some aggregation necessary to activate the pyrogenic process in rabbits as well as activate the coagulation pathway for the LAL assay. Highly purified monomeric LPS molecules do not retain biological activity. An E. coli LPS was chosen as the RSE as it represented the “middle of the road” pyrogenic activity. The RSE (second generation) is currently used by LAL manufacturers to standardize their secondary CSE standards and is in limited supply. A description of RSE/CSE standardizations was no longer officially in print when the original FDA “blue book,” Guideline on Validation of the LAL Test as an End-Product Endotoxin Test for Human and Animal Parenteral Drugs, Biological Products, and Medical Devices, (1987), was retired. The RSE/CSE standardization procedures were not included in the compendial chapters for the Bacterial Endotoxin Test (BET).
The use of RSE worldwide by lysate vendors allowed the BET test to be performed globally using one worldwide standard. Therefore, an endotoxin unit (measure of endotoxin activity) from the US is the same as any other EU/IU worldwide. A more detailed description of the RSE, CSE and NOE can be found in a recently published Pharmacopeial Forum article8. The article includes the nature of LER, LLR and proposed ways to mitigate the instability of CSE versus stable NOE..
Much data has been generated demonstrating that LER is really LLR. Bolden and colleagues9 have demonstrated that NOE activity is stable in chelating citrate/PS80 buffers while CSE exhibited the LLR phenomenon. My experiments10 performed using NOE (E. cloacae) versus CSE also confirmed Genentech’s findings for CSE loss of activity while the activity of NOE remained stable regardless of the container type or temperature of storage. Attempts to reverse the CSE to an active state were unsuccessful. Dubczak11 extended experimentation of the NOE to several GNB sources verses the RSE standard to include concurrent LAL and RPT. The data indicated that NOE was easily recovered over time and pyrogenic in vivo while the RSE activity was pyrogenic at T=0 but lost its activity both in the rabbit and in vitro LAL testing by 24 hours or storage at room temperature. The activity was 50 EU/mL and 1mL per kg was dosed. See Figures 1 for Time = 0 and Figure 2 for T=24 hours.
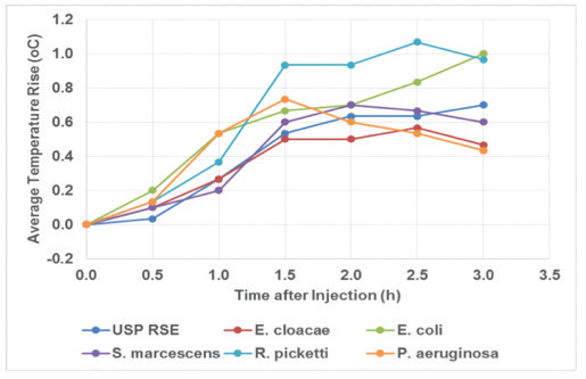 Figure 1.
Figure 1.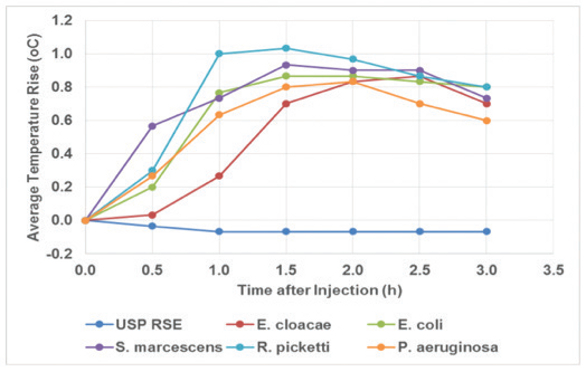 Figure 2.
Figure 2.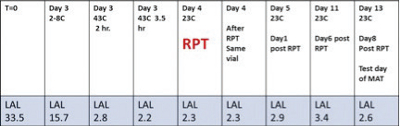 Figure 3. Theoretical CSE activity added: 30 EU/mL
Figure 3. Theoretical CSE activity added: 30 EU/mL
RPT Dose: 1mL per kg
RPT temperature rise results: 0.1, 0.4, 0.2 °C
MAT results: 1 EU/mLI also performed CSE versus RPT and MAT which demonstrated a loss of activity of the CSE in a 20mM citrate/0.025% PS matrix. This matrix with surrogate CSE failed to be pyrogenic in the RPT and MAT tests. The activity levels for the data shown in Figure 3 would have been well above that required to elicit pyrogenic responses and LAL/MAT activity values. Therefore, the LAL test is an accurate and reliable predictor of true endotoxin. A loss of activity as measured by the LAL and MAT tests correlates with a loss of pyrogenic activity in vivo during rabbit pyrogen testing.
The FDA has expressed concerns about the variability of the in vivo and in vitro test results being submitted to the agency for new biological products12. One has to also understand the variability of the LAL test methodologies, the insensitivity of the RPT, and the importance of a robust and reliable experimental design to interpret and understand the variability of the results.
One source of variability of the data is the methodology chosen to perform the assays. The gel clot test is a semi-quantitative sensitivity titration test that relies on a physical gel formation diluted to its endpoint. This assay cannot accurately quantitate endotoxin activity loss when small amounts of analyte are being added directly to product matrices. The variability of the gel clot assay likely exceeds the accuracy necessary to determine percent activity losses. The apparent activity loss can also be observed in the kinetic methods if low levels of surrogate analyte are added to the product matrix coupled with a dilution necessary to dilute out matrix interferences. The results may appear as a loss of endotoxin when one has actually lost the sensitivity of the assay’s ability to discern the differences between test interference and LPS activity.
Another source of data variability can be in the interpretation of the % loss of activity. It is important to perform pyrogen free water positive control results to assess activity losses. Data has demonstrated that for some matrices, the loss of LPS is almost immediate. Analyzing data against a water control is the most accurate evaluation of the data. In this manner one can assess day to day assay variability.
Experimental design can also play a large role in the variability of the RPT. Incorrect assumptions have been made that an endotoxin activity of 5 EU/kg will cause pyrogenic reaction in rabbits. Greisman and Hornick13 conducted parallel pyrogen tests in both humans and rabbits using endotoxin from Salmonella typhosa, Escherichia coli, and Pseudomonas species. Their results indicate that rabbit pyrogenic responses (temperature rises) peak at 1.5 hours after injection, while those in man peak at approximately 3 hours. They also determined that the threshold pyrogenic doses for rabbits and man are similar. It has been demonstrated that the degree of aggregation of the endotoxin molecules correlate with the degree of in vitro pyrogenicity. The less aggregated the endotoxin molecules are, the less pyrogenic they become. In the studies previously mentioned as well as a few other historical studies, the endotoxin from S. typhosa was the most potent followed by the E. coli endotoxin. Lagging far behind in pyrogenicity was endotoxin from Pseudomonads which is more likely than E. coli to be present in our industry. In reality the 5 EU/kg endotoxin threshold currently used for calculating endotoxin limits has a 2 to 10 fold safety margin. Rabbits that had been given 10 EU/kg of endotoxin passed the RPT 90% of the time. Some rabbits passed the RPT at 80-500 EU/kg while most rabbits passed the RPT at 1-40 EU/kg. Rabbits inoculated with Pseudomonas derived endotoxin did not have pyrogenic responses until they received 1200 to 2500 EU/kg. In an article by Dr. Hochstein et al14, E. coli derived LPS was tested extensively in rabbits. An IV dose of 10 to 15 EU/kg was necessary to produce even a minimal (0.5C or higher) pyrogenic reaction. Therefore, pyrogenicity observed at levels less than 10 EU/kg on three rabbits is highly suspect. Injections of rabbits with protein products have been known to cause fever responses in rabbits due to factors other than endotoxin activity. It is important to remember that rabbits that have been exposed to human proteins for these tests cannot be reused whether or not they exhibited pyrogenic responses during the RPT and are usually euthanized.
Endotoxin is the clinically relevant analyte. Most modern products prepared in cGMP facilities where the raw materials, water, facilities, personnel, or manufacturing processes are highly controlled often contain no measurable endogenous endotoxin. Many endotoxin specifications for biological product have been set far below the endotoxin calculations using the 5 EU/kg and dose per hour criteria set forth in the USP. Dr. Fred Pearson15 reported that while highly purified endotoxin preparations should be used as LAL standards, they should not serve as an absolute standard of pyrogenicity because they do not parallel the pyrogenicity of environmental endotoxins. Marlys Weary along with Dr. Pearson, et al16 stated that environmental endotoxin is consistently less pyrogenic than LPS standards probably because in our industry, most (endotoxins) are Pseudomonads and not E. coli. It is important that industry and regulatory agencies understand more clearly the relationship of the rabbit and the LAL test systems with regard to environmental endotoxin.
Conclusions
Understanding the history and development of the LAL test and it’s correlation to the RPT helps us to understand the true nature of LPS as CSE and RSE versus the activity of NOE. While the LPS standards serve to provide an activity measurement against which pass or fail acceptance criteria are measured, the demonstration of the stability of assayable endotoxin should be performed on assayable natural endotoxin. Natural endotoxin should be used as a surrogate for demonstrating the stability of assayable endotoxin in the absence of endogenous analyte in the product. Other attempts to assay the stability of LPS or to re-aggregate LPS to its active form are purely academic and nonvalue added as LPS does not exist naturally.
References
- Guidance for Industry Pyrogen and Endotoxins Testing: Questions and Answers, U.S. Food and Drug Administration. June 2012.
- Chen, J. “Low Endotoxin Recovery in Common Biologics Products.” Presented at the 2013 PDA Annual Meeting. Orlando, FL, April 2013.
- Levin J, Bang FB. The role of endotoxin in the extracellular coagulation of Limulus blood. Bull Johns Hopkins Hosp 1964;115:265-274.
- Cooper J, Levin J, Wagner HN. Quantitative comparison of in vitro (Limulus) and in vivo (rabbit) methods for the detection of endotoxin. J. Lab. Clin. Med. 1971; 78:138-148.
- Bowers K, Tran, L. Creation of an in-house naturally occurring endotoxin preparation for use in endotoxin spiking studies and LAL sample hold time analysis. American Pharmaceutical Review. 2011;14(6):92-97.
- Westphal O, Luderitz O. Uber die extraktion von bakterien mit phenol/wasser. A. Naturforsch. 1952 7b: 148-155.
- Rudbach JA, Akiya J, Elin RJ. Preparation and properties of a national reference standard endotoxin. J. Clin. Microbiol. 1976; 3:21-25.
- Bolden J, Platco C, Dubczak J, Cooper J, McCullough KM. The use of endotoxin as an analyte in biopharmaceutical product hold time studies. Published in Pharmacopeial Forum, 2015.
- Bolden JS, Clarebout ME, Miner MK, et al. Evidence against a bacterial endotoxin masking effect in biologic drug products by Limulus amebocyte lysate detection. J Parenter Sci Technol. 2014;68(5):472-477.
- Platco C. Low lipopolysaccharide recovery versus low endotoxin recovery in common biological product matrices. American Pharmaceutical Review, 2014;17(Endotoxin Detection Suppl Part II):4-7.
- Dubczak J. A comparative in vitro and in vivo low endotoxin recovery (LER) assessment. PDA Annual Global Conference on Pharmaceutical Microbiology. Bethesda, MD, 2014.
- Hughes PF, Thomas C, Suvarna K. et al. Low endotoxin recovery: an FDA perspective. BioPharma Asia. 2015;4(2). www.biopharma-asia.com.
- Greisman S.E. and Hornick R.B., Proc.Soc.Exp.Biol. and Med., Vol. 131:1154 (1969)
- Hochstein HD, et al. the processing and Collaborative Assay of a Reference Standard. Journal of Biological Standardization (1983) 11:251-260.
- Pearson, F. The Pyrogenicity of Environmental Endotoxins and LPS Endotoxins and Their Detection with the Limulus Amebocyte Lysate Test, Progress in Clinical and Biological Research, Vol. 93:261 (1982) Alan R. Liss, Inc.
- Weary, Pearson, et al. The Activity of Various Endotoxins in the USP Rabbit Test and in Three Different LAL Tests. Endotoxins and Their Detection with the Limulus Amebocyte Lysate Test,pg 365-379. 1982 Alan R. Liss, Inc.