Large Scale Production of Biologics is Susceptible to Microbial Contamination
The term “Biologics” is used for a class of therapeutics that are produced by recombinant DNA technology and generally fall into three major categories: (i) therapeutic proteins, (ii) monoclonal antibodies (mAbs), and (iii) antibody-drug conjugates.
Large scale production of biologics consists of two process steps, which often occur at different production sites: (i) Drug Substance (DS) manufacture (manufacture of the Active Pharmaceutical Ingredient), and (ii) Drug Product (DP) manufacture (manufacture of the Final Drug, e.g. formulated mAbs filled in vials or syringes). In the DS manufacturing process bacterial or mammalian cells are used to express the desired recombinant protein. Following cell expansion and production phases these cultures are harvested, the recombinant protein is recovered and is purified to separate the target protein from host cells, cell debris, and process- and host-related impurities. The purified protein is then formulated and filtered into containers for storage and shipment to the DP manufacturing site where the formulated DS is then filled into vials or syringes to produce the final drug product. In the DP process most biologics are subject to terminal sterilization as they are administered parenterally (e.g. intravenous, subcutaneous, intravitreal). However, for biologics manufacturing the risk of microbial contamination is not limited to the final product. Many biologics manufacturing steps (e.g. protein purification, conditioning, formulation) occur under non-sterile conditions in aqueous systems at ambient temperature or 2-8 °C under substantially neutral pH conditions, making the large-scale production of biologics susceptible to microbial contamination (see Figures 1 and 2 for a typical mAb production process). Regardless of where in the DS or DP process they occur, microbial contaminations can have a significant impact on product quality and patient safety. Microbial contaminations can lead to product degradation or modification due to the introduction of microbial enzymes. In addition, the introduction of soluble components derived from the microorganisms can adversely impact patient safety. The investigation of microbial excursions must adequately assess impact to both product quality and patient safety prior to making decisions regarding lot release.
 Figure 1. Typical process flow for DS manufacturing of therapeutic mAbs.
Figure 1. Typical process flow for DS manufacturing of therapeutic mAbs. Figure 2. Typical process flow for DP manufacturing of therapeutic mAbs.
Figure 2. Typical process flow for DP manufacturing of therapeutic mAbs.Development of a successful microbial control strategy for largescale production of biologics should be based on a risk assessment to ensure end-to-end microbial control of the process through adequate prevention and detection. The risk assessments require review of the microbial quality of direct materials, utilities (WFI, process gases, etc.), and individual steps within the series of unit operations that constitute the DS and DP manufacturing process. For each step, the assessment should evaluate process/operational conditions and existing controls regarding the equipment, the manufacturing environment, and personnel preventive controls for their effectiveness at controlling and minimizing microbial contamination, with a goal of prevention of ingress, and proliferation in the process, equipment, and product. In addition, the existing microbial detection controls should be assessed to ensure they provide continued verification that the microbial prevention controls are working as intended. Detection controls consist of bioburden and endotoxin testing against established limits on samples obtained from defined process steps. For critical process steps (e.g. production culture and DS) the acceptable bioburden limit is extremely low and valid bioburden results exceeding acceptance criteria would lead to rejection of the batch.
Microbial Risk of Biologics Manufacturing is Not Limited to Living Microorganisms and Intact Microbial Cells
The active pharmaceutical ingredient in biologics products are usually heat sensitive. Therefore, filtration using membranes with pore sizes ranging from 0.02 to 0.2 µm size is the method of choice for bioburden reduction and terminal sterilization. While 0.02 to 0.2 μm filtration can effectively remove intact microbial cells from the product, this process does not allow removal of subcellular structures of bacteria and fungi. If co-purified with the product subcellular structures like microbial toxins and so called Pathogen Associated Molecular Pattern (PAMPs), i.e. endotoxins, lipopeptides, lipoproteins, flagellin, bacterial and fungal DNA, and cell wall polysaccharides potentially lead to toxic, allergic, or inflammatory responses in humans. In addition, copurified extracellular proteases or endoglycosidases potentially lead to product degradation or modification.
In other words, bioburden contaminations of non-sterile process intermediates represent a risk even after 0.02 to 0.2 μm filtration, and even if both Drug Substance and Drug Product specifications are met. This risk is also described in relevant guidelines:
USP“Monitoring of Bioburden”:
“Bioburden is a potential risk to the patient not only because the sterilization process might not be completely effective, but also postprocessing because of the possible presence ofresidual materials such as allergens, endotoxins, and exotoxins. It may also have adverse impact on product quality and stability.”
FDA, Questions and Answers on cGMP (January 2011):
“What manufacturing contamination risks are presented by the different pathogenic agents ?
… Microbial toxins can be divided into two general groups: exotoxins and endotoxins. ….. Exotoxins, especially heat-stable exotoxins, can remain in the ingredient throughout the manufacturing process and adversely affect patient health…….. Some species of molds produce toxic byproducts called mycotoxins. ….”
Bioburden and Endotoxin Testing Play a Key Role in The Assessment of Microbial Risk of Non-Sterile Process Intermediates
The microbial detection control strategy of large scale biologics production defines bioburden and endotoxin sampling points for nonsterile process intermediates. Endotoxin testing using the compendial LAL assay allows universal detection of bacterial endotoxins, while direct detection of other toxins and non-endotoxin PAMPs derived from bioburden in process intermediates is a challenge, making it difficult to reliably confirm their presence or absence. Bioburden analysis performed on representative samples before 0.2 μm filtration (so called pre-filtration samples) allows determination of the total microbial load of the product and serves as a surrogate test for toxins and nonendotoxin PAMPs. The potential load of toxins and non-endotoxin PAMPs can be calculated based on the bioburden concentration (e.g. CFU/10 mL), cell characteristics of the contaminating organism, process step, and microbiological factors of toxins and non-endotoxin PAMPs. The calculated load can be compared to safety limits specific for toxins and non-endotoxin PAMPs.
Bioburden analysis can also indicate the potential presence of degradative enzymes, e.g.; extracellular proteases, or endoglycosidases, which could cause product degradation or modification. In some cases stability studies can be conducted to assess the potential impact of these microbial components on product quality.
Case-by-Case Assessment of Bioburden (CCAB): A Comprehensive Approach to Assess the Impact of Bioburden Contaminations of NonSterile Process Intermediates on Product Quality and Patient Safety.
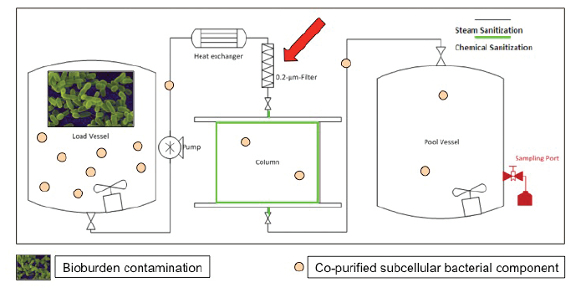 Figure 3. Example for microbial contamination event during Biologics Manufacturing. A column load tank is contaminated with bacteria (illustrated as rods). Living bacteria and intact bacterial cells are removed from the product by the 0.2 µm filter (red arrow). However, subcellular bacterial components pass the 0.2 µm filters and can be co-purified with the product.
Figure 3. Example for microbial contamination event during Biologics Manufacturing. A column load tank is contaminated with bacteria (illustrated as rods). Living bacteria and intact bacterial cells are removed from the product by the 0.2 µm filter (red arrow). However, subcellular bacterial components pass the 0.2 µm filters and can be co-purified with the product.The following sections describe the Case by Case Assessment of Bioburden (CCAB) procedure and underlying rationales. The CCAB procedure is used to investigate bioburden action limit excursion in the Roche/Genentech Biologics manufacturing network and is only one component of the quality investigation. The investigation must be reviewed holistically and all findings that may impact product quality, patient safety, equipment fitness and process performance must be considered before determining acceptability of the batch. The applicability of CCAB is assessed by quality based on the criticality of the impacted process and whether there are mechanisms for additional removal of impurities derived from bioburden in subsequent process step(s).
Figure 4 illustrates the key aspects of CCAB, which are described in more detail below.
 Figure 4. Key aspects of CCAB approach.
Figure 4. Key aspects of CCAB approach.Patient Safety Assessment (PSA)
General Aspects
Patient safety assessment focuses on subcellular (extracellular) components of bacteria and fungi, which show the following characteristics:
- components potentially pass filters installed in the manufacturing process to reduce bioburden or viral loads (0.02 – 0.2 μm pore size).
- components cannot be detected using analytical methods, which are established in a routine GMP QC environment.
- components are PAMPs, allergens or exotoxins relevant to human safety.
Subcellular/extracellular components of bacteria and fungi, which show all aforementioned characteristics, are termed “critical components”. Patient safety assessment procedure is used to determine the patient safety impact based on hypothetical assumptions about the amount and potency of critical components in a microbial load. The assumption is that critical components will be co-purified into the Drug Substance and the finished pharmaceutical product (DP).
Critical components comprise the following subcellular microbial structures:
Protein exotoxins
Bacteria are capable of producing a wide range of protein toxins most of them are actively released from bacterial cells during growth and therefore are considered as exotoxins (Alouf, 2000). Examples for bacterial protein exotoxins are hemolysins and staphylococcal enterotoxins (Dingens et al., 2000). Production of hemolysins is also reported for fungal species like Aspergillus fumigatus (Wartenberg et al., 2011) and Candida spp. (Luo et al., 2001). Several protein exotoxins like staphylococcal enterotoxins and toxic shock syndrome toxin 1 can act as classical allergens (Novak et al., 2003). Currently, over 300 different protein exotoxins are described (Alouf, 2000). In contrast to bacterial endotoxins, which are universally detected by the LAL assay a wide range of different test methods exists for bacterial protein toxins (Pimbley and Patel, 1998). Most protein exotoxins occur as single-chain holoproteins varying from approximately 2-3 kDa to approximately 300 kDa (Alouf, 2000). As a worst case scenario it is assumed that protein exotoxins are not excluded by 0.02 – 0.2 µm filters.
Non-protein exotoxins
Fungal species are capable of producing a wide range of non-protein exotoxins also referred to as mycotoxins (Bennett and Klich, 2003). No universal test method exists, which detects all mycotoxins (Turner et al., 2009). For bacteria only a few non-protein toxins are described. Tetrodotoxin (TTX or puffer fish toxin) is one of the most potent neurotoxins. It is produced by different bacterial strains covering a wide taxonomic range (e.g. strains of the genera Pseudomonas, Bacillus or Microbacterium) (Lu and Yi, 2009). Some mycobacteria produce mycolactones, which are suspected to cause Buruli ulcer, an unusual skin disease (Hong et al., 2008). A variety of bacterial strains from different bacterial genera (Pseudomonas, Burkholderia, Acinetobacter, and Rhodococcus) have been shown to produce rhamnolipids as LPSlike exotoxins (Andrä et al., 2006). As a worst case scenario it is assumed that non-protein exotoxins are not excluded by 0.02 – 0.2 µm filters.
MALP-2 like lipopeptides/proteins
Lipopeptides/proteins are peptides or proteins modified by covalent linkages to lipids, with various functions in the bacterial cell. They are widespread in Gram-negative bacteria, but are also found in Gram-positive bacteria. The overwhelming majority of lipopeptides/proteins are anchored to the cell membrane (Narita et al., 2004; Hutchings et al., 2008). Of relevance to CCAB are lipopeptides/proteins similar to MALP-2 lipopeptide (a 2 kDa macrophage-activating lipopeptide) from Mycoplasma fermentans that are found in the extracellular protein fraction. MALP-2 has been shown to have an inflammatory potency similar to that of LPS (Galanos et al., 2000). Since bacterial lipopeptides can be shown to be endotoxic on account of their chemical structure (Schromm et al., 2007), they can be assumed to have a potency essentially comparable to that of MALP-2 or LPS. Based on the current literature fungal lipopeptides/proteins do not act as PAMPs (Sorrell and Chen, 2009). Lipopeptides/proteins vary greatly in size. As a worst case scenario it is assumed that they are not excluded by 0.02 – 0.2 µm filters.
Flagellin
Flagellin is the protein component of the bacterial flagellum, the most common organelle for locomotion of Gram-positive and Gramnegative bacteria. Flagellar filaments and monomers are shed into the environment (Neish, 2007). Flagellin is a potent inducer of inflammatory responses (Neish, 2007). Approximately 20,000 flagellin subunits are present in a single flagellar filament of 20 nm diameter and 10-15 µm length (Hughes and Erhardt, 2011). Depending on the bacterial species, flagellins have molecular masses ranging from 28 to 80 kDa (Winstanley and Morgan, 1997). Given these numbers it is highly unlikely that single flagellin subunits are size excluded by 0.02 – 0.2 µm filters.
Bacterial and fungal DNA
Release of plasmids and chromosomal DNA has been shown for Bacillus subtilis during growth (Lorenz et al., 1991). As a worst case scenario it is assumed that the majority of bacteria and fungi are capable of actively releasing DNA. Bacterial DNA induces inflammatory responses in humans because of the high frequency of unmethylated CpG motifs (Krieg, 2002; Akira et al., 2006). Unmethylated CpG motifs has also been found in Aspergillus fumigatus DNA (Ramirez-Ortiz et al., 2008). As a worst case scenario it is assumed that unmethylated CpG motifs are also abundant in other fungal species. It is highly unlikely that 0.02 – 0.2 µm filters exclude immunostimulating microbial DNA since CpG motifs are also present on short DNA stretches or fragmented DNA.
Cell wall polysaccharides
Polysaccharides are structural elements of bacterial and fungal cell walls. Microbial cell wall synthesis is a highly dynamic process. As a worst case scenario it is assumed that cell wall polysaccharides are shed into the environment. For Gram-positive bacteria peptidoglycan (PGN) constitute 30-70% of the dry weight of the cell wall (Schlegel, 1985). The cell wall of Gram-positive bacteria contains lipoteichoic acid (LTA), which is absent in Gram-negative bacteria. Both LTA and PGN are potent inducers of inflammatory responses (Rockel and Hartung, 2012). For Gram-negative bacteria endotoxins and lipoproteins constitute up to 80% of the dry weight of the cell wall, whereas PGN constitutes only a small fraction of less than 10% (Schlegel, 1985). The fungal cell wall is 80-90% polysaccharides with most of the remainder consisting of protein and lipid. The most important structural elements of the fungal cell wall are chitin and ß-glucan (Bartnicki-Garcia, 1968). As a worst case scenario it is assumed that cell wall polysaccharides or fragments of it are not excluded by 0.02 – 0.2 µm filters.
PSA Procedure
Evaluation of Microorganisms
The starting point for the compilation of a PSA is a quantifiable microbial contamination. Identification of the representative microorganisms in the contamination uses ribosomal DNA sequencing technique.
Situation A:
The microorganism is identified to genus or species level. Information about critical components and cell characteristics which might impact patient safety is obtained through literature searches. Literature searches should cover but are not restricted to:
- Presence of exotoxins relevant to human safety
- Presence of MALP-2 like lipopeptide/proteins
- Presence of flagellin
- Cell wall type (gram-positive, gram-negative, fungal cell wall)
- Cell size (bacteria and yeasts) or size of conidia (molds)
Exceptions include microorganisms that for methodological reasons cannot be unambiguously assigned to a species or genus based on ribosomal DNA sequences, but for which the presence of toxins can be assumed due to their taxonomic affiliation (e.g. members of the Enterobacteriaceae family).
Situation B:
The taxonomic affiliation of the recovered microorganism is unknown as it cannot be identified to species, genus or family level. It is assumed that microorganisms, which cannot be identified to genus or species level by ribosomal DNA sequences do not produce exotoxins relevant to humans. It is current scientific standard that ribosomal DNA sequences of microbial isolates including those of clinical relevance are deposited in public databases like GenBank/EMBL or SILVA1 . Therefore it can be excluded that exotoxin producing microorganisms recovered from a contamination cannot be identified to genus or species level by ribosomal DNA sequencing2 . Additional investigations (e. g. Gramstaining, test for motility etc.) are needed to assess critical components other than exotoxins.
Calculation of Hypothetical Loads of Critical Components
Microbiological factors for calculating hypothetical loads of critical components are given in Table 1.
Table 1. Microbiological factors for calculating hypothetical loads of critical components.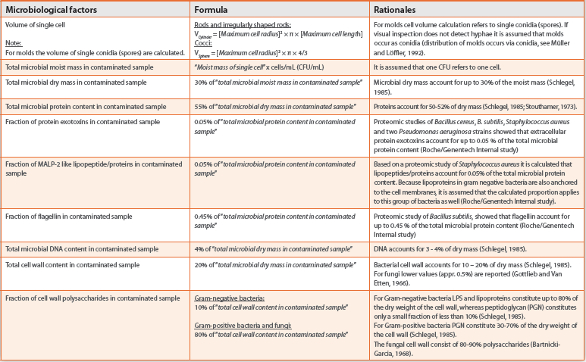
Calculation of Carryover of Critical Components
For the calculation of the hypothetical carryover of critical components into the DS and the DP, a distinction is made between downstream processing and upstream processing.
Upstream processing
Upstream processing comprises the process steps from fermentation or cell culture through harvest including cell separation and clarification, and where applicable, inclusion body preparation/renaturation (E. coli processes). A distinction must be made between cell culture products and E.coli products.
Cell culture products:
The protein active substances (e.g. monoclonal antibodies) are secreted into the cell culture medium. Any critical component present is able to bind non-specifically to the proteins and thereby be carried over into down stream processing. The assumption is made here that 10% of the critical component binds (non-specifically) to the protein active substances3 .
E. coli products:
Nonspecific binding of any critical component present to the protein active substance (in the form of inclusion bodies or as periplasmic proteins) is possible only after cell disruption. The assumption is made here that 10% of the critical component binds (nonspecifically) to the inclusion bodies/to the refolded or the periplasmic protein active substance and is thereby carried over into down stream processing for E. coli products. Critical components bound to the E. coli cell wall/cell surface are removed during cell disruption/clarification and cannot thereby be carried over into downstream processing. Carryover of critical components, by this route, into the DS or into the DP can therefore be ruled out.
Downstream processing
Downstream processing comprises the process steps from loading onto the first chromatography column up to bulk compounding and formulation. In the event of microbial contamination in downstream processing, complete carryover of the critical component into the bulk active substance and the finished pharmaceutical product is assumed for in-process samples. No distinction is made between “early” and “late” process steps.
Calculation of Critical Component Dose
The pharmaceutical product is assumed to be administered at a certain dose per day. The calculation of the hypothetical dose of critical component exposed to humans is based on the product concentration of the corresponding drug substance solution. If the product concentration is not available, the lowest product concentration value defined by the specification is used. The number of doses per mL can be calculated from the maximum dosage of the drug. This information can be obtained from product inserts or leaflets.
Assessment of Risk Potential
Safety limits for critical components are given in Table 2. A worst case body weight should be selected based on the description for each product or information obtained from clinical trials. If no documentation is available, 50 kg body weight for absolute dosing (non-weight-based) or dosing per patient should be selected to represent worst case scenario.
Table 2. Risk potential assessment of critical components.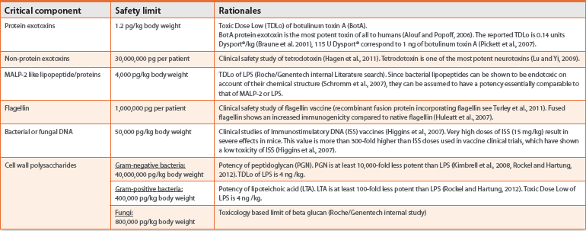
Product Quality Impact Assessment (PQIA)
General Aspects
Co-purified extracellular proteases, endoglycosidases or other microbial enzymes can potentially lead to product degradation or modification, impacting product quality. To further investigate this hypothesis microbial proteomics of a panel of microorganisms that have been recovered from Biologics product streams and known to produce extracellular proteases and other modifying enzymes (Bacillus cereus, Pseudomonas aeruginosa, Stenotrophomonas maltophilia, and Staphylococcus aureus) was conducted (Roche/Genentech internal study, Figure 5 illustrates the experimental setup).
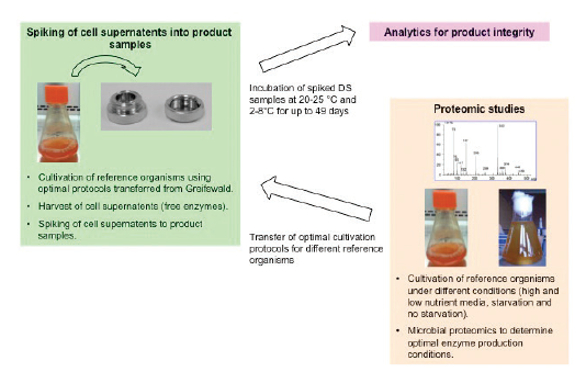 Figure 5. Experimental setup of proteomic study to evaluate the impact of extracellular microbial enzymes on product stability.
Figure 5. Experimental setup of proteomic study to evaluate the impact of extracellular microbial enzymes on product stability.Proteomic analysis of microbial cell supernatants showed that release of those enzymes is strongly influenced by growth conditions (high and low nutrient media, starvation and no starvation). Cell supernatants were taken from panel microorganisms grown under optimal enzyme production conditions, spiked into two different Biologics (one therapeutic mAb and one key signaling protein) and incubated at 20-25 °C and 2-8 °C for up to 49 days. After incubation spiked product samples were analyzed for product integrity. In some cases significant changes of product integrity were seen even when spiked cell supernatants resulted from cell counts as low as 100 CFU/mL.
The PQIA procedure considers these study results. A review and evaluation of relevant QC data provides an indicator whether there is impact to product quality. In certain cases, additional product stability testing is required to determine impact.
PQIA procedure
The PQIA differentiates between process steps with potential for subsequent reduction of modifying or degrading components derived from bioburden, and process steps for which no further reduction of modifying or degrading components would be expected.
Process steps with further downstream purification
For all steps of upstream processing and all downstream process steps up to the load of the last process step with potential reduction of modifying or degrading components, PQIA is performed as follows:
- Assess relevant CoA data (e.g., identity, potency tests) of the impacted drug substance or drug product lot.
- Compare data against reference materials or other released lots for potential anomalies.
Process steps with no further downstream purification
For all process steps where further reduction of critical components derived from bioburden is not expected (e.g. final UFDF, thawed bulk, bulk compounding, and drug product processing steps) the PQIA is performed as follows:
- Assess relevant CoA data (e.g., identity, potency tests) of the impacted drug substance or drug product lot.
- Compare data against reference materials or other released lots for potential anomalies.
- Perform a product stability study under protocol using the product stability indicating assays for the product. Appropriate subject matter experts must assess any anomalous data for impact to product quality.
Summary
Large scale production of biologics is susceptible to microbial contamination. Bioburden contaminations of non-sterile process intermediates represent a risk to patient safety and product quality. Even after bioburden removal by 0.2 µm filtration, and even if both Drug Substance and Drug Product specifications are met, subcellular microbial components like toxins, lipopeptide/lipoproteins, flagellin, bacterial and fungal DNA, cell wall polysaccharides, extracellular proteases or endoglycosidases remain in the product. Those microbial components potentially lead to toxic, allergic or inflammatory responses in humans or product degradation or modification. The CCAB approach described here enables a comprehensive assessment of these risks.
Acknowledgement
The author would like to thank Ray Arnold, Wolfgang Eder, Emabelle Ramnarine for helpful comments on the manuscript and Sven Deutschmann, Holger Kavermann, Michael Knight, Christian John, Markus Haberger, Ingo Lindner, Andreas Adler, Andreas Krug, Prof. Ulrich Zähringer (Research Center Borstel, Germany), Prof. Uwe Völker and Elke Hammer (University of Greifswald, Germany), for their contributions to develop the CCAB concept.
References
- Latest version of SILVA database (released September 2016, see http://www.arb-silva.de) contains over 6,300,000 ribosomal DNA sequences.
- An exception are microorganisms that for methodological reasons cannot be unambiguously assigned to a species or genus based on ribosomal DNA sequences, but for which the presence of toxins can be assumed due to their taxonomic affiliation (e.g. members of the Enterobacteriaceae family).
- This is a worst case assumption based on an internal process capability studies of endotoxin, CHO proteins, and CHO DNA removal.
- Akira et al., 2006. Cell. 124:783-801.
- Alouf and Popoff. 2006. “The comprehensive Sourcebook of Bacterial Protein Toxins”. 3rd edition. Elsevier Ltd.
- Alouf, 2000. Bacterial Protein Toxins: An Overview, in: Bacterial Toxins. Methods and Protocols. Holst O, ed., Walker, J. M. Humana Press, Totowa, NJ, USA, pp. 1-27.
- Andrä et al., 2006. Biol. Chem. 387:301-310.
- Bartnicki-Carcia. 1968. Annu. Rev. Microbiol. 22:87-108.
- Bennett and Klich. 2003. Clin. Microbiol. Rev. 16:497-516.
- Bhatia and Zahoor, 2007. J. Clin. Diagn. Res. 3:188-197.
- Braune et al., 2001. British J. Dermatology. 144:111-117.
- Dingens et al., 2000. Clin. Microbiol. Rev. 13:16-34.
- Galanos et al., 2000. J. Endotoxin Res. 6:471-476.
- Gottlieb and Van Etten. 1966. J. Bacteriol. 91:161-168.
- Hagen et al., 2011. Curr. Oncol. 8:e109-e116.
- Higgins et al., 2007. Expert Rev. Vaccines 6:747-759.
- Hong et al., 2008. Nat. Prod. Rep. 25:447-454.
- Hughes and Erhardt. 2011. eLS. John Wiley & Sons, Ltd: Chichester.
- Huleatt et al., 2007. Vaccine. 25:763-775.
- Hutchings et al., 2008. Trends in Microbiol. 17:13-21.
- Kimbrell et al., 2008. Immunol. Lett. 118:132-141.
- Krieg. 2002. Annu. Rev. Immunol. 20:709-760.
- Lorenz et al., 1991. Arch. Microbiol. 156:319-326.
- Lu and Yi, 2009. Annals of Microbiology. 59:453-458.
- Luo et al., 2001. J. Clin. Microbiol. 39:2971-2974.
- Müller and Löffler. 1992 Mykologie, 5. Aufl., Thieme Verlag, Stuttgart
- Narita et al., 2004. Arch. Microbiol. 182:1-6.
- Neish. 2007. Am. J. Physiol. Gastrointest. Liver Physiol. 292:462-466.
- Novak et al., 2003. J. Allergy Clin. Immunol. 112:215-216.
- Pickett et. al., 2007. European J. Neurologia. 14:E11.
- Pimbley and Patel. 1998. J. Appl. Microbiol. Biotechnol. 84:98-109.
- Ramirez-Ortiz et al., 2008. Infection Immunity. 76:2123-2129.
- Rockel and Hartung. 2012. Frontiers in Pharmacology. 3:1-19.
- Schlegel, H. G. (1985), Allgemeine Mikrobiologie, 6. Aufl., Thieme Verlag, Stuttgart.
- Schromm et al., 2007. J. Biol. Chem. 282:11030-11037.
- Sorrell and Chen. 2009. Adv. Exp. Med. Biol. 666:108-20.
- Stouthamer. 1973. Antonie van Leeuwenhoek. 39:545-565.
- Turley et al., 2011. Vaccine. 29:5145-5152.
- Turner et al., 2009. Anal. Chimica Acta. 632:168-180.
- Wartenberg et al., 2011. Int. J. med. Microbiol. 301:602-611.
- Williams et al., 1988. Int. J. Immunopharmac. 10:405-414.
- Winstanley and Morgan. 1997. Microbiology. 143:3071-3084.