Abstract
Potential Genotoxic Impurities (PGIs) and Genotoxic Impurities (GIs) have to be controlled to low parts per million levels in active pharmaceutical ingredients and drug products. Method development and analysis of PGIs/GIs at these trace levels present unique challenges to the analytical chemist. In this work, we present a comprehensive strategy with decision trees to guide the method development process for the analysis of these PGIs/GIs. Depending on the physicochemical properties of the particular PGIs/GIs and the required sensitivity, analytical chemists can choose appropriate techniques such as Gas Chromatography (GC) and Liquid Chromatography (LC) coupled with different detectors. Some case studies of PGIs/GIs analysis are illustrated.
Introduction
Synthesis of Active Pharmaceutical Ingredients (APIs) is a multi-step process involving the use of reactive chemicals, reagents, solvents, catalysts, and salts. Residual levels of process-related impurities, byproducts, and degradants could have adverse health effects. To ensure patient safety, global regulatory agencies require API manufacturers to reduce the levels of such compounds to safe levels. International Conference on Harmonization (ICH) documents Q3A(R2) and ICH Q3B(R2) provide guidance on limiting the majority of these less toxic impurities in new drug substances and drug products respectively.1,2 However, some reactive Genotoxic Impurities (GIs) even when present at very low levels could potentially bind to the DNA or proteins leading to gene mutation. The European Medicines Agency (EMA) and the Food and Drug Administration (FDA) issued guidance documents that highlight the importance of this issue and have mandated limits and controls GIs in drug substances. ICH issued draft guidance in February 2013 which provided a framework for identification, categorization, qualification, and control of Potential Genotoxic Impurities (PGIs). As per ICH M7 guidance, reagents, starting materials, intermediates, byproducts, process-related impurities, and potential degradation products in drug substances are categorized into classes 1 through 5 based on comprehensive hazard evaluations commonly known as in-silico assessments. Known mutagens (Class 1 and Class 2) and compounds with structural alerts (Class 3) need to be controlled at low parts per million levels as recommended in ICH M7.3-5
The major challenges encountered in the analysis of PGIs/GIs are the wide ranges in the volatility, polarity, and reactivity of these compounds which is further compounded by differences in sample matrices ranging from starting materials to intermediates to APIs. The quantitation limits for PGIs/GIs are usually several orders of magnitude lower than less toxic impurities nominally 0.05%. The specification/ acceptance criteria are usually set to low parts per million levels based on duration of exposure (acute vs. chronic) and drug product dosage strength making it extremely challenging to analyze and quantitate these compounds.
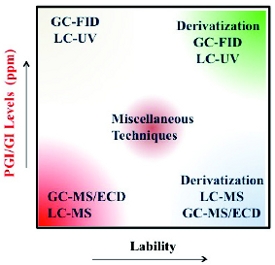 Figure 1. A general strategy for selecting the appropriate analytical technique for PGIs/GIs analysis.
Figure 1. A general strategy for selecting the appropriate analytical technique for PGIs/GIs analysis.A good understanding of the stability and reactivity of PGIs/GIs as well as their specification limits is critical in selecting the appropriate technique for their analysis as shown in Figure 1. In general, chromatographic techniques like Liquid Chromatography (LC) and Gas Chromatography (GC) are used to analyze stable compounds whereas labile compounds would often require derivatization prior to LC or GC analysis. For some selected analyses, Liquid Chromatography- Ultraviolet Detection (LC-UVD) and Gas Chromatography-Flame Ionization Detection (GC-FID) techniques may be deemed suitable. However, Liquid Chromatography-Mass Spectrometry (LC-MS) and Gas Chromatography-Mass Spectrometry (GC-MS) are usually the techniques of choice when higher specificity and sensitivity are required. Similarly, Gas Chromatography-Electron Capture Detection (GC-ECD) techniques are commonly used for halogenated PGIs/GIs to enhance sensitivity and selectivity. Occasionally, some spectroscopic techniques like Nuclear Magnetic Resonance (NMR), light scattering, and Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) are used in analyzing PGIs/GIs (see Figure 1, Miscellaneous techniques).
Method Selection for PGIs/GIs Analysis
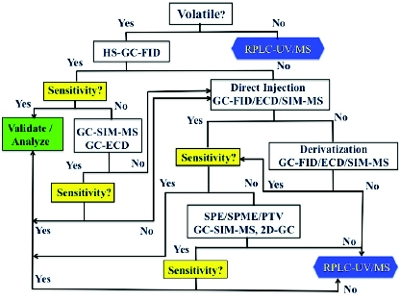 Figure 2. Decision tree for volatile PGIs/GIs analysis.
Figure 2. Decision tree for volatile PGIs/GIs analysis.Analytical approaches based on physicochemical properties of PGIs/GIs have been proposed in the literature and are better alternatives to developing individual assays for these compounds.6 However, the decision trees presented are very general and lack details for novice analysts.
In this work, we present a comprehensive strategy for the analysis of PGIs/GIs taking into consideration the physicochemical properties of the compounds of interest and the required specificity and sensitivity of the analytical techniques as shown in Figures 2 and 3. The choice of detectors is mainly dictated by the required limits that need to be reached while demonstrating lack of matrix interference.

Figure 3. Decision tree for non-volatile PGIs/GIs analysis.
Figure 2 shows the decision tree for volatile compounds. For volatile PGIs/GIs, headspace GC with FID detection is usually the technique of choice.7 The sample matrix containing the API and its related impurities is usually highly soluble in organic diluents and does not interfere with the analysis due to the low partitioning into the headspace (low vapor pressure). The high solubility and low vapor pressures would allow for low detection limits. Typically, if the desired sensitivity is obtained, the method is finalized and validated for further analysis. If higher sensitivity is required, other detectors like ECD or MS8 can be used.
Semi-volatiles are often analyzed using direct injection GC with FID, ECD, or Single Ion Monitoring-Mass Spectrometry (SIM-MS) detection depending on the required sensitivity. Caution needs to be applied with direct injection analysis of PGIs/GIs as potential degradation of concentrated samples in the GC inlet system can contaminate the GC system and result in false positives. ECD (for halogenated compounds) or SIM-MS detection are sometimes critical in improving sensitivity and specificity of direct injection analysis.
An application of direct injection GC-MS analysis for GIs is shown in Figure 4. Nitro compounds are usually used as precursors in the synthesis of API starting materials and are usually genotoxic in nature. Although these compounds are relatively small, the presence of chloro, fluoro substituents reduces their volatility precluding the use of headspace GC. SIM-MS enhances the sensitivity and selectivity of direct injection analysis, enabling successful analysis of nitro impurities at detection limits as low as 1 ppm.
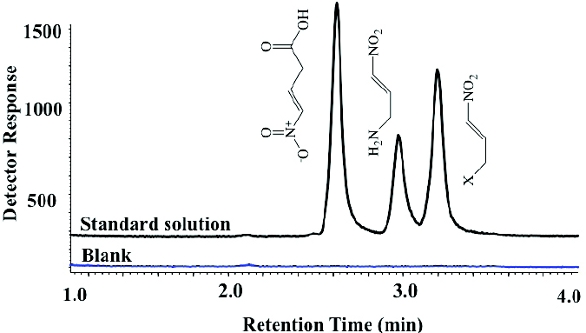 Figure 4. GC-MS analysis of nitro compounds. SIM offers sensitivity and specificity for genotoxic impurities with minimal interference from the sample matrix.
Figure 4. GC-MS analysis of nitro compounds. SIM offers sensitivity and specificity for genotoxic impurities with minimal interference from the sample matrix.If the sensitivity is still not reached for the desired analysis, techniques like Solid Phase Micro-Extraction (SPME), Solid Phase Extraction (SPE), Programmed Temperature Vaporization (PTV), and Two-dimensional- Gas Chromatography (2D-GC) can often enhance sensitivity and specificity of PGIs/GIs analysis by selectively extracting/transferring components of interest thereby minimizing the matrix effects. Sample derivatization9-11 is typically conducted for non-volatile or reactive PGIs/GIs to improve their volatility and stability prior to analysis by GCFID/ ECD/MS.
For non-volatile compounds, Reversed Phase HPLC (RPLC) coupled to a UV or MS detector is the technique of choice as illustrated in Figure 3. For PGIs/GIs containing chromophores, RPLC-UV can be chosen as long as the method can demonstrate specificity and sensitivity. If successful, the method can be finalized and validated for further analysis. Additional sensitivity can be obtained by performing stacked injections or using a MS detector for ionizable compounds.
For RPLC-UV methods that do not demonstrate suitable specificity, Mixed-mode Chromatography (MMC) or Hydrophilic Interaction Liquid Chromatography (HILIC) can be used. These two techniques can significantly improve the retention of very polar compounds. As always, the MS detector can enhance the sensitivity and selectivity if needed.
Genotoxic impurities with ionic functional groups are difficult to chromatograph by RPLC due to their poor interaction with hydrophobic stationary phases. Ion pairing reagents are often used to enhance their retention. Mixed mode chromatography with acidic and/or basic functional groups embedded in hydrophobic stationary phase provides an easier alternative eliminating the need for ion-pairing reagents. Application of mixed mode chromatography for the analysis of aminopyrazole, a genotoxic starting material in an API sample is shown in Figure 5. Impact of the modifier and its strength as well as the stationary phase on the sensitivity and specificity of aminopyrazole is presented. Aminopyrazole, a basic compound, is well retained in a Primesep-200 column (pKa approximately 2) under acidic conditions as shown in Figure 5B. Increasing the strength of formic acid from 0.05% to 0.1% results in a decreased retention of aminopyrazole as shown in Figure 5C. However, the retention of benzene sulfonic acid eluting around 1 minute is not impacted (Figure 5B and Figure 5C). Switching to a stronger eluent (phosphoric acid instead of formic acid) results in a further decrease in retention of aminopyrazole as shown in Figure 5D along with an increase in signal-to-noise (S/N) ratio. However, retention of benzene sulfonic acid increases with the use of phosphoric acid (present in the fully protonated form). Moving to a weaker ion-exchange stationary phase, Primesep-C (pKa ~ 3), results in elution of aminopyrazole in column pre-void due to its repulsion of the stationary phase (Figure 5E). This results in further enhancement in S/N ratio. Therefore, by changing the acidic modifier and its strengths, stationary phase, the retention of polar ionic compounds is readily manipulated in mixed mode chromatography improving sensitivity and selectivity. Overall, a 6-fold increase in sensitivity was observed for aminopyrazole.
 Figure 5. Analysis of aminopyrazole, a basic genotoxic impurity using mixed mode chromatography. Depending upon the acidic modifier, its strength, and stationary phase, significant enhancement in sensitivity and selectivity can be achieved.
Figure 5. Analysis of aminopyrazole, a basic genotoxic impurity using mixed mode chromatography. Depending upon the acidic modifier, its strength, and stationary phase, significant enhancement in sensitivity and selectivity can be achieved.For non-volatile compounds lacking chromophores, RPLC/MS could be used to provide adequate specificity and sensitivity. As mentioned earlier for molecules containing chromophores, improving specificity can be established by performing MMC or HILIC chromatography. Stacked injections can enhance the sensitivity further to achieve extremely low limits of detection.
Reactive, labile compounds can be converted to more stable HPLC amenable species using derivatization techniques.12,13Figure 6 shows an example of PGI analysis using esterification as a derivatization technique. Acid chlorides are key intermediates in API synthesis due to their inherent reactivity and coupling capability. However, many of these compounds are PGIs and readily hydrolyze to their corresponding acids which are non-genotoxic. Hence, the analysis of these compounds is very challenging. In this example, acid chlorides were converted to stable methyl ester using anhydrous methanol under ambient conditions. The methyl ester eluting in the proximity of API is difficult to analyze using UV detection. However, since the [M + H]+1 ion of the ester is unique, it enables selective ion monitoring of the esters off ering desired specificity and sensitivity. HPLC analysis of the reactive acid chloride as the methyl ester in unspiked and spiked API samples using UV and MS detection is shown in Figure 6. As shown, detection limits as low as 2 ppm can be achieved.
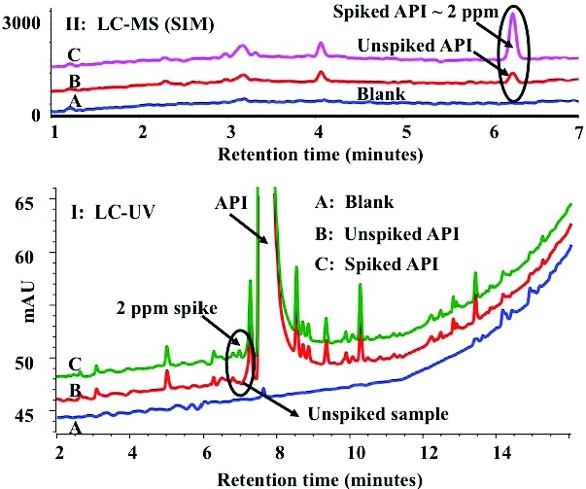 Figure 6. HPLC analysis of reactive acid chlorides as methyl esters via derivatization with anhydrous methanol. The LC-UV trace of diluent blank, unspiked, and spiked API is shown at the bottom (I) and LC-MS trace is shown at the top (II). SIM offers sensitivity and specificity with minimal interference from other chemical components.
Figure 6. HPLC analysis of reactive acid chlorides as methyl esters via derivatization with anhydrous methanol. The LC-UV trace of diluent blank, unspiked, and spiked API is shown at the bottom (I) and LC-MS trace is shown at the top (II). SIM offers sensitivity and specificity with minimal interference from other chemical components.In cases where all the aforementioned techniques do not work, Twodimensional- Liquid Chromatography (2D-LC-MS) can be investigated. In 2D-LC-MS, peaks co-eluting on primary column are resolved on a complementary secondary column minimizing potential interference from sample components. An application of 2D-LC-MS in the analysis of residual ethyl besylate, a Genotoxic Impurity is shown in Figure 7. Ethyl besylates could be formed by the reaction of ethanol with benzene sulfonic acid during the synthesis of API. An overlay plot of diluent blank, a 5-ppm standard of ethyl besylate, unspiked and spiked API samples (10 mg/mL) are shown at the bottom in Figure 7A. Ethyl besylate co-elutes with the API in the primary phenylhexyl column making it difficult to analyze by UV and or MS detection. A fraction of this co-eluting component when transferred to a complementary secondary C18 column resolves ethyl besylate from the API enabling UV and MS detection as shown in the overlay plots at the top in Figure 7B and Figure 7C. MS offers better specificity compared to UV detection and would allow for lower detection limits if needed.
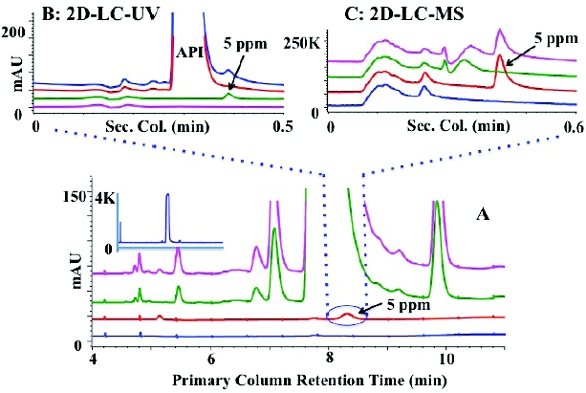 Figure 7. 2D-LC-MS separation of a PGI (ethyl besylate). Figure A shows the primary column separation on a phenylhexyl column with ethyl besylate co-eluting with the API. Figure B and Figure C show UV and MS traces of the secondary C-18 column. The co- eluting ethyl besylate is resolved from the API in the secondary dimension.
Figure 7. 2D-LC-MS separation of a PGI (ethyl besylate). Figure A shows the primary column separation on a phenylhexyl column with ethyl besylate co-eluting with the API. Figure B and Figure C show UV and MS traces of the secondary C-18 column. The co- eluting ethyl besylate is resolved from the API in the secondary dimension.Conclusions
We presented a strategy to guide pharmaceutical analytical chemists during the development of analytical methods for low level PGIs/GIs quantitation. Depending on the structure of the PGI/GI and desired limit of detection, one can easily use these decision trees to define the appropriate method. For volatile PGIs/GIs, headspace or direct injection GC-FID, GC-ECD, or GC-MS can be investigated. Additionally, sensitivity can be greatly enhanced by using SPME, SPE, or PTV with GC-MS or 2DGC techniques. For non-volatile PGIs/GIs, RPLC-UV or RPLC-MS can be investigated. Selectivity can be enhanced by performing MMC or HILIC chromatography, and sensitivity can be easily improved by stacked injections. For volatile or nonvolatile compounds, derivatization can be employed to change the physicochemical properties of the PGIs/ GIs to allow for suitable analysis. For extremely challenging analysis, 2D-GC or 2D-LC-MS can be used to demonstrate superior selectivity and sensitivity.
Acknowledgements
The authors would like to thank Larry Wigman, James Girotti, Ila Patel, Emily VanHassel, Christine Gu, Kavita Mistry, Nik Chetwyn, Minli Xie, Remy Angelaud, Mark Reynolds, and David Stirling from Genentech Inc., for their contributions.
Author Biographies
C. J. Venkatramaniis a senior scientist at Genentech, USA and has over fifteen years' experience in the pharmaceutical industry. He was the key member of Genentech technical team instrumental in taking gRED’s first small molecule Erivedge, from development to commercial. Erivedge, is currently approved in several countries around the world for the treatment of advanced BCC. His areas of interests include ultra-trace analysis and multi-dimensional chromatography.
Mohammad A. Al-Sayah, PhD,got his doctorate in Analytical Chemistry from Florida State University in 2004. He then joined the Analytical Chemistry department at Merck & Co as a Senior Research Chemist. In 2012, Mohammad became a scientist in the Small Molecule Analytical Chemistry group at Genentech where he is a scientific group leader providing analytical support for drug substance and drug product development.
References
- International Conference on Harmonisation. Q3A(R2): Impurities in New Drug Substances. 2006.
- International Conference on Harmonisation. Q3B(R2): Impurities in New Drug Products. 2006.
- Guideline on the Limits of Genotoxic Impurities, Committee for Medicinal Products (CHMP), European Medicines Agency, London, 28 June 2006. (CPMP/SWP/5199/02, EMEA/CHMP/ QWP/251344/2006).
- EMEA (European Medicines Agency) “Question & Answers on the CHMP Guideline on the Limits of Genotoxic Impurities”, adopted 26 June 2008.
- International Conference on Harmonisation. M7: Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk. 2013.
- Teasdale A. Genotoxic Impurities: Strategies for Identification and Contol. Hoboken, NJ: John Wiley & Sons, Inc.; 2010.
- Pierson DA, Olsen BA, Robbins DK, DeVries KM, Varie DL. Org. Process Res. Dev. 2009;13:285.
- Ramjit HG, Singh MM, Coddington AB. J. Mass. Spectrom. 1996;31:867.
- Regis Chromatography Catalog. 1998-1999.
- Knapp DR. Handbook of Analytical Derivatization Reactions. New York, NY: John Wiley and Sons; 1979.
- Blau K, King G. Handbook of Derivatives for Chromatography. London, UK: Heyden & Sons Ltd.; 1979.
- Lunn G, Hellwig LC. Handbook of Derivatization Reactions for HPLC. New York, NY: John Wiley and Sons; 1998.
- Lawrence JF, Frei RW. Chemical Derivatization in Liquid Chromatography. Journal of Chromatography library. Volume 7. Elsevier Scientific Publishing Company; 1976.