Introduction
Over the last couple of decades, antibody-drug conjugates (ADCs) have continued to evolve as a rapidly growing drug targeting technology for the treatment of cancer. ADCs consist of a cytotoxic drug connected by a linker to an antibody, which delivers it specifically to the target cancer cells with abundant cell surface associated antigens1. This mechanism, thus, restricts the cell killing activity of cytotoxic drugs to the cancer cells and improves patient’s safety2.
This review article focusses on the unique properties and the related challenges of ADCs at their conjugation, manufacturing, stability and analytical testing levels.
DAR, Unconjugated Antibody, and Free Drug at the Conjugation Stage
The development of a commercially viable ADC is contingent on a robust conjugation process, capable of ensuring that the critical quality attributes unique to ADCs, such as drug antibody ratio (DAR), unconjugated antibody, and free drug content, be within phase appropriate limits at release and during the intended use period by the patient. During the pharmaceutical product development of an ADC, these attributes must be demonstrated to be consistently within the proposed acceptance criteria from early phase development through commercialization, despite the expected associated changes in manufacturing scale up, equipment or manufacturing site change.

The DAR and the relative percentages of the various drug-loaded ADC species are extremely important for an ADC as they can impact its binding, potency, clearance, toxicity, and stability3. A low DAR implies reduced ADC yield and a retarded ADC efficiency due to competition from lower conjugated or unconjugated antibody molecules. However, ADC species having the same DAR measurement can be positional isomers as well, differing in their conjugation position of the light or heavy chains of the antibody. Such positional isomers can be different in their in-vitro properties, as well as in their in-vivo profiles3,4. The precise control of DAR and drug distribution becomes more complex for lysine linked ADCs relative to cysteine conjugated ones. For lysine linked ADCs, conjugation can theoretically take place on any of the 40 unique solvent exposed lysine residues per antibody by direct acylation or with hetero-bi-functional cross-linking agents resulting in one million possible ADCs5. On the other hand, cysteine linked conjugation takes place by alkylation at the eight thiol groups produced by the reduction of four interchain disulfide bonds, resulting in ADC species having a DAR of 0, 2, 4, 6 or 85,6. The average DAR and the drug distribution of an ADC should be monitored at the conjugation level through tight in-process controls to ensure batch to batch consistency.
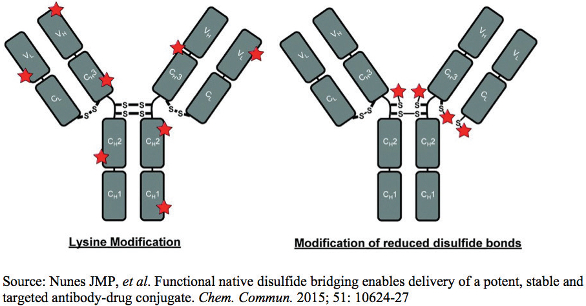 Figure 1. Random Conjugation Chemistry of ADCs
Figure 1. Random Conjugation Chemistry of ADCsThe concept of unconjugated or free drug is very critical to an ADC, since any fraction of the conjugated drug released prior to reaching the intended tumor site is unavailable for the desired therapeutic effectiveness. On the other hand, this free drug is capable of causing serious side effects to the patient owing to its highly cytotoxic nature, and possible formation of long-circulating adducts with endogenous proteins such as albumin7.
The unconjugated antibody content should be minimal at the release of an ADC batch. The applied conjugation process is expected to utilize the entire antibody content available for conjugation. Any unconjugated antibody fraction will eventually compete with the ADC for the same antigens at the tumor site, without producing any therapeutic benefit to the patient.
Essentially, any change in conjugated antibody or conjugated drug content will impact the DAR and the relative percentages of the various drug loaded ADC species available to the patient. The fraction of free drug and unconjugated antibody should be controlled through the conjugation process to ensure consistency at the release of a batch, and through its shelf life for the efficacy and safety of an ADC.
Site-specific conjugation technologies employing engineered cysteine residues, unnatural amino acids, and enzymatic reactions with trans-glutaminases are being investigated to have better control over the DAR and the drug distribution of an ADC, by circumventing the heterogeneity associated with lysine or cysteine linked conjugation techniques5,8. However, ADCs prepared employing site specific conjugation techniques are yet to be commercialized.
Toxicity of ADCS at the Manufacturing Stage
The manufacture of ADCs often needs dedicated suites for handling these raw materials, owing to their high toxicity, especially from the cytotoxic drug component. Appropriate cleaning procedures must be in place to prevent cross contamination of these highly potent products.
Stability and the Role of Analytical Testing
ADCs like any biologic may experience con-ditions compromising their stability, ranging from shear stresses during their manufacturing in the ultra-filtration/ diafiltration procedures, formulation in buffers, storage, temperature excursions during shipping and handling or even during use by the patients. ADCs are routinely monitored for their purity, size variants, charge variants, DAR, unconjugated antibody, and free drug content throughout their intended shelf-life. A clear understanding of the degradation pathways of ADCs is essential to recommend their anticipated storage conditions and to support their shelflife statement.
In general, the stability of an ADC is partially determined by the stability characteristics of its unconjugated antibody. However, the stability of antibodies has been reported to be negatively impacted following conjugation with cytotoxic drugs, as evidenced by a reduced melting point (Tm) with increasing DAR9. The conjugation of a hydrophobic cytotoxic drug to an antibody can favor antibody unfolding through its binding to the hydrophobic amino acid residues normally buried in its interior or by altering the higher order structures of the antibody. It is a well-known fact that the higher the DAR, the greater the aggregation tendency of an ADC. Moreover, many of the currently available linkers such as Val-Cit and Phe-Lys linkers are relatively hydrophobic in nature, and can induce aggregation of ADCs10. The purity of an ADC can be established by size exclusion chromatography (SEC), although SEC can be limited for the determination of soluble reversible aggregates, due to dilution of the samples prior to analysis, as well as from subsequent dilution in the mobile phase. Organic modifiers need to be added judiciously to the mobile phase, without altering the native structure of the ADCs and the higher molecular weight species, to overcome the non-specific interactions between the stationary phase and the hydrophobic cytotoxic drug11. ADC fragments, disulfide cross linkages, and the covalent or non-covalent nature of the aggregates can be evaluated by capillary electrophoresis-sodium dodecyl sulfate (CESDS), particularly for lysine-linked conjugates. ADCs conjugated at their cysteine residues may not be analyzed under the denaturing conditions of the CE-SDS method in the presence of disrupted covalent interchain disulfide bonds, and the possible ready fragmentation of the antibodies in the presence of SDS.
Table 1. Common Analytical Tests Employed for ADCs on Stability

ADCs can undergo chemical degradations like deamidation, glycation, N-terminal pyroglutamation to form acidic charge variants. On the other hand, they can form basic charge variants from chemical disulfide exchange of the free thiol groups from unpaired cysteine residues intermolecularly and intramolecularly, oxidation or succinimide formation. The charge profile of an ADC is dependent on the net charges on the antibody, the cytotoxic drug, and the conjugation chemistry. The possibility of conjugation to the epsilon amino group of any of the 40 unique lysine residues on an antibody molecule can result in a complicated analysis of heterogeneous population of charge variants. This is supplemented by the fact that the conjugation of cytotoxic drugs reduces the net positive charge of the antibody by one unit for each lysine residue conjugated3,12. Such a phenomenon interferes with the inherent surface electrostatic properties of the antibody, making the determination of charge variants of an ADC challenging by conventional ion exchange chromatographic techniques. Isoelectric focusing (IEF) separations can be very complex for the determination of charge variants for ADCs owing to the charge heterogeneity of lysine linked conjugates.


The DAR of an ADC may change on storage as a result of deconjugation, thus impacting the efficacy and the safety profile of the drug product. DAR can be measured by UV spectrometry, given the antibody and the cytotoxic drug absorbing UV light at different wavelengths, so that the concentrations of the drug and the antibody can be independently calculated. However, the employed UV method is not capable of distinguishing the absorbance of the conjugated drug from the free form, and may lead to an overestimated DAR if the amount of free drug is not subtracted from the total amount of measured drug. The comparatively mild and non-denaturing hydrophobic interaction chromatography (HIC) is particularly suitable for the determination of average DAR, the free drug, and the drug distribution for cysteine linked ADCs11. Reverse phase high performance liquid chromatography (RP-HPLC) analyses under reducing condition can provide information on the amount of free drug and the average DAR of cysteine linked ADCs. However, RP-HPLC may not be applied for the determination of DAR of ADCs linked by lysine conjugation, owing to the highly heterogeneous nature of the resulting ADC species produced13. Mass spectrometry (MS) is a useful orthogonal method to confirm the average DAR and the relative percentage of the drug loaded species as it is insensitive to the complexity resulting from the presence of a very large number of positional isomers. The main limitation to the determination of DAR by MS is that it assumes a uniform response for all of the conjugated species which may not always be the case. However, the DAR determined by MS is usually with 5-10% of the value obtained by UV. The determination of sites of conjugation is a significant analytical challenge, especially for lysine-conjugated ADCs due to the large number of potential conjugate sites. The most widely reported approach used is the analysis of the reduced and proteolytically digested ADC using HPLC coupled with high resolution MS.
Conclusions
The science of ADCs is growing rapidly as a drug targeting technology for the treatment of cancer. However, the development and commercialization of ADCs often encounters multiple challenges at their conjugation, manufacturing, and stability levels owing to their unique properties such as DAR, unconjugated antibody, free drug or even due to their high toxicity. Often orthogonal analytical methods are required to be employed to resolve these challenges and provide a commercially viable ADC.
References
- Wang Z, et al. Antibody-Drug Conjugates: The Forefront of Targeted Chemotherapy for Cancer Treatment. J Drug Des Res. 2015; 2(3): 1016-24.
- Chari RVJ, et al. Antibody-Drug Conjugates: An Emerging Concept in Cancer Therapy. Angew Chem Int Ed. 2014; 53(15): 3796-3827.
- Wakankar A, et al. Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs. 2011; 3(2): 161-72.
- Drake PM, Rabuka D. An emerging playbook for antibody-drug conjugates: lessons from the laboratory and clinic suggest a strategy for improving efficacy and safety. Curr Opin Chem Biol. 2015; 28: 174-80.
- Panowksi S, et al. Site-specific antibody drug conjugates for cancer therapy. MAbs. 2014; 6(1): 34–45.
- Drachman JG, Senter PD. Antibody-drug conjugates: the chemistry behind empowering antibodies to fight cancer. Hematology Am Soc Hematol Educ Program. 2013; 2013: 306-10.
- Tumey LN, et al. In vivo biotransformations of antibody-drug conjugates. Bioanalysis. 2015; 7(13): 1649-64.
- Goswami S, et al. Developments and Challenges for mAb-Based Therapeutics. Antibodies 2013; 2(3): 452-500.
- Beckley NS, et al. Investigation into temperatureinduced aggregation of an antibody drug conjugate. Bioconjug Chem. 2013; 24(10): 1674-83.
- Dubowchik GM, et al. Doxorubicin immunoconjugates containing bivalent, lysosomallycleavable dipeptide linkages. Bioorg Med Chem Lett. 2002; 12(11): 1529-32.
- Yi Du, et al. Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. mAbs. 2012; 4(5): 578–85.
- Boylan NJ, et al. Conjugation Site Heterogeneity Causes Variable Electrostatic Properties in Fc Conjugates. Bioconjug Chem. 2013; 24(6): 1008–1016.
- Stephan JP, et al. Anti-CD22-MCC-DM1 and MC-MMAF conjugates: impact of assay format on pharmacokinetic parameters determination. Bioconjug Chem. 2008. 19(8): 1673-83.
Acknowledgement
The author wishes to express her sincere thanks to Dr. Mark Bolgar and Dr. Mark Arnold from Bristol-Myers Squibb for critically reviewing this manuscript and providing their valuable comments.
Author Biography
Nila Das is a Senior Research Investigator at the New Brunswick R&D site of Bristol-Myers Squibb in New Jersey. Her research interests include therapeutic monoclonal antibodies and antibody-drug conjugates. Most recently she had led the development of parenteral products at Boehringer Ingelheim. She obtained her Ph.D. in Industrial Pharmacy from St. John’s University in 2008.