Introduction
Over the last decades drug development expertise for biologicals has constantly increased. Nevertheless failures still happen and are mostly related to intolerable toxicity, low efficacy and undesired physicochemical and biopharmaceutical attributes. A complete overview can be found in Figure 1. Although overall the success rates for biologicals are a little better in comparison to small molecules, just the attrition rate could be further reduced by a more systematic approach, assessing the “developability” of new biological entities (NBE) at an early stage. This strategy positively impacts cost, time and quality. Firstly, it is favorable to know as much as possible about the molecule as early as possible to avoid any negative surprises at a more costly time point. This includes also the consideration whether the protein can be manufactured at production scale at competitive costs. Secondly, speed to market must be taken into account. Any frontloading of activities that result in an earlier access to the market are worth millions as the patent lifetime is limited. Thirdly, improved quality will contribute to better patient satisfaction and therapeutic efficacy. Therefore developability assessment is introduced to enable a ranking of several potential product candidates not only based on their biological activity. This article describes typical strategies to predict the behavior of molecules in terms of aggregation, stability, potency, pharmacokinetics and -dynamics (PK/PD), solubility, viscosity, immunogenicity and safety, taking into account sample consumption, duration of the assays and amenability to high throughput. These methods have usually been established for monoclonal antibodies, however most of them are applicable to any other NBE as well. This supplies scientists in drug development with an arsenal of tools which can be utilized beyond standardized platforms that work only for certain molecule classes. Although most companies have specific strategies to cover developability aspects,1 it can be observed that the majority utilizes the tools at two distinct points in the drug development pathway. An overview of the typical elements can be seen in Figure 2.
 Figure 1. Potential sources of failures during development (in red: parameters assessed for developability)
Figure 1. Potential sources of failures during development (in red: parameters assessed for developability) Figure 2. Developability assessments at the discovery/development interface
Figure 2. Developability assessments at the discovery/development interfaceSelection and Optimization Phase
Usually the first entry point is during the initial candidate screening and selection phase, when more than 10 molecules are available that display sufficient biological activity in terms of affinity, selectivity and reactivity. In case the molecules are derived from genetic libraries, an in silico sequence analysis is performed. Here critical parameters such as the isoelectric point (pI), charge distribution, aggregation-prone regions (APR), positions for post translational modifications (PTM) potential recognition sites for proteases and the epitope based risk for immunogenicity are evaluated. Information about the pI is useful for designing appropriate downstream processing (DSP) steps and selecting suitable excipients for formulation. Charged amino acids should be distributed across the molecule and not concentrated into large patches that might lead to aggregation or adsorption on biological tissues. PTM sites are quite relevant as in general unpaired cysteines should not be present to avoid disulfide scrambling or intermolecular crosslinking leading to aggregation. Recognition sequences for N-linked glycosylation to asparagine residues being sources for heterogeneity or regions susceptible to proteolytic degradation should be omitted. On the other hand if these amino acids are required for biological function such as proteolytic activation or to enable a certain glycan pattern for molecular recognition, they should be at the correct position and properly modified. APRs in therapeutic proteins are identified by measuring the dynamic exposure of hydrophobic patches. If using homology modeling or working with a static structure, predictions become less accurate but can be obtained much faster.2 The immunogenicity risk can be predicted by looking for T- or B-cell epitopes that elicit either a cellular or humoral immune response. Unfortunately it is almost impossible today to predict B-cell epitopes as they, in contrast to linear T-cell epitopes, have a conformational dependency. B-cell epitopes cause the generation of anti-drug antibodies (ADA) that can either neutralize the activity of the drug, alter the pharmacokinetics or even worse, cross-react with autologous proteins.3
In case the protein is not derived from a library, obvious sequence and structure liabilities should already be eliminated during molecule design. This includes removal of free cysteines, or other solvent exposed amino acids sensitive to oxidation as methionine or tryptophan. Furthermore, asparagine residues are critical as they are the anchor point for N-linked glycosylation and tend to isomerize to aspartic acid or just like glutamine to deamidation. Also the presence and position of lysine must be scrutinized, as it can be the target for glycation, the covalent conjugation of reducing sugars to the free amine group. However, when dealing with non-antibody proteins, homology modelling of structures gets difficult as probably not enough information on structural conformation is available in databases.
For the next steps protein material is required. This is usually generated by transient expression in human embryonic kidney (HEK) cells which can deliver the quantities for subsequent high throughput (HTP) assays. During the candidate selection phase information about aggregation behavior, experimental hydrophobicity and stability is desirable. The easiest assessment of aggregation can be achieved by size exclusion chromatography (SEC) followed by multi angle laser light scattering (MALLS). This powerful method can also identify low molecular weight species resulting from degradation or misassembled fragments in the case of antibodies and their derivatives. The presence of exposed hydrophobic patches is determined by retention in standard hydrophobic interaction chromatography (HIC). Stability can indirectly be evaluated through differential scanning fluorimetry (DSF) that measures the fluorescence of a dye when bound to hydrophobic regions in a protein at different conditions.4
The information gained from in silico analysis and the experimental data can now be used either to re-engineer and mutagenize the protein of interest5 or to eliminate candidates with unfavorable properties.
After the first round of molecule assessment during lead optimization within discovery, the next step is beyond the interface within the phase of preclinical development. Typically no more than four top lead candidates remain that have been improved or chosen based on the information from the selection phase. The first step now is to generate stable cell lines to express the proteins of interest. The selection of the best cell line is based on parameters such as doubling time of approximately 24 h, a cell concentration of more than 1×107 mL-1, a protein output of more than 10 pg per cell and day, a cell stability beyond 20 passages and an average viability above 95%. For all further experimental investigations not necessarily clonal cells are required, sometimes pool material is sufficient. This is particularly true for cells generated by site directed integration with relatively uniform properties instead of random integration with hugely variable results.
Now material for the following more extensive studies must be supplied. The initial approach is a standard cultivation of cells in shake flasks delivering harvest for the initial capture step. In the case of antibodies this is relatively straight forward as affinity chromatography with protein A resins exists. All other proteins must rely on specific properties such as charge and pI to enable high capacity ion exchange chromatography. The first eluate from the capture step should already have a purity above 70% to be applicable for subsequent more detailed stability studies simulating potential extreme and nonphysiological conditions during the process. This includes exposure to elevated temperatures, freeze-thawing cycles, storage in a wide range of pH values, and mechanical stress such as pumping, filtration or mixing. Of course, oxidative stress and the influence of light that could degrade aromatic amino acids and the peptide backbone should not be neglected.6 The sensitivity towards proteases in body fluids can be assessed by incubation in human or primate serum. In general it is advised to distinguish between structural and colloidal stability. One of the suitable methods is standup monolayer adsorption chromatography (SMAC). Retention times on this column type are inversely related to colloidal stability. As it is essentially a SEC analysis, further important properties as monomer content or solubility can be assessed simultaneously, thus saving time and sampling.7
Formulation
Today formulation development is increasingly important as many drugs are administered subcutaneously at high concentrations (>100 g L-1) and low volumes (High viscosity is often caused by protein self-interaction that depends on non-uniform charge distribution with positive and negative patches. This can be partly predicted by in silico models, but the experimentally determined values from dynamic light scattering (DLS) with inert latex beads are more reliable for non-antibody molecules. Solubility is indirectly deducted from experiments with HIC and aggregation studies but nowadays also HTP viscometer instruments are available.9
At the initial stage, a simple formulation might be sufficient to get through early development phases. However, it is recommended to complete a more thorough optimization as early as possible to have a final formulation that that is suitable up to commercialization.
Strategic Recommendation
Developability assessments typically are installed at the interface between discovery and development.10 They serve the purpose of selecting the right candidate(s) to enter the development stage. At this point the desired biological attributes are established, so other parameters can be selected to identify the ideal candidate. Most companies follow a two-tier approach that facilitates the optimization of a limited number of leads after the first round. This selection phase combines in silico sequence analysis with several HTP assays supplied by small scale transient expression.
A handful of top candidates successfully passing that first filter will then be used to generate stable pools and ultimately stable cell lines. These cell lines are then the sources for material supply to support all further studies comprising extensive stability assessments under non-physiological conditions and paving the way for proper up- and downstream process development. Formulation is a special case as it benefits from the information gathered in both phases, but might be fully established only after finalizing the manufacturing process thus stretching into the clinical development. It is a thin line between performing a full blown formulation development too early and wasting money and effort on failing candidates or to invest too late, thus loosing valuable time.
As a lot of data is accumulated about many different candidates it is recommended to integrate the information into IT solutions that enable a systematic ranking as schematically displayed in Figure 3.
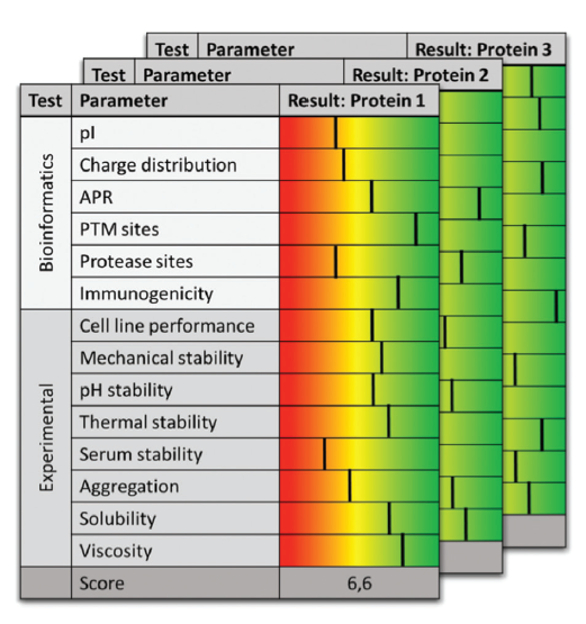 Figure 3. Ranking matrix for developability assessment
Figure 3. Ranking matrix for developability assessmentThe overall goal is to integrate discovery (lead selection and optimization activities) with process development by implementing methods that facilitate risk assessment with minimal material requirements and maximal throughput. The focus is on balancing efforts not to spend too much early, but sufficiently to move forward with a suitable candidate. Interestingly, different strategies can be observed in different company types. For instance small and young companies being cash flow and time constrained might tend to limit the efforts in order to reach only the next financial inflection point for subsequent fund raising. Established companies on the other hand are more willing to invest more early on in order to have a solid understanding which candidate to pursue further. Contract manufacturing organizations, often not involved in candidate selection, still can benefit from this holistic approach to initiate process development, as developability studies deliver a modular tool kit to address typical reoccurring issues.
References
- Jarasch, A. et al. J. Pharm. Sci. 104, 1885–1898 (2015).
- Chennamsetty, N. et al. J. Phys. Chem. B 114, 6614–6624 (2010).
- De Groot, A.S. et al. Curr. Opin. Pharmacol. 8, 620–626 (2008).
- Senisterra, G.A. et al. Mol. BioSyst. 5, 217–223 (2009).
- Seeliger, D. et al. MAbs 7, 505–515 (2015).
- Hawe, A. et al. J Pharm Sci 101, 895–913 (2012).
- Kohli, N. et al. MAbs 7, 752–758 (2015).
- Yang, Y. et al. Biotechnol. Bioeng. 114, 2043–2056 (2017).
- Deshmukh, S. et al. ACS Comb. Sci. 18, 405–414 (2016).
- Yang, X. et al. MAbs 5, 787–794 (2013).