Abstract
The aim of this investigation is to compare the release behavior of a multiple-unit modified-release formulation (‘tablets in capsule’) of Mesalazine, with dextran as excipient, against powder filled capsules in various gastrointestinal simulated pH media. The results indicate that capsules, when filled with minitablets, release.
Introduction
The anti-inflammatory drug Mesalazine is mainly used in the symptomatic treatment of Ulcerative Colitis, Crohn’s disease and inflammatory bowel disease. Mesalazine exerts its action locally in the inflamed area of the intestine and not through systematic absorption. Campregher and Gasche (2011)1 have reported that when Mesalazine is administered orally, it dissolves in the stomach, and therefore, cannot act therapeutically in the targeted colon area. For this reason, researchers have designed novel Mesalazine carrier systems, which act in the colon either by the use of specific excipients, or creating pro-drugs, or by changing the route of administration (i.e.: enema, suppository).
Lopes and his colleagues (2006)2 have shown that the per os administration of controlled release formulations can be either single unit dosage forms (SUDFs), e.g. tablets and capsules, or multiple unit dosage forms (MUDFs), like granules, pellets and minitablets. MUDFs offer an efficient control of drug release, because the total dose is administered in many single units. Advantages of the MUDFs include: smaller inter- and intra-variability, higher degree of dispersion in the gastrointestinal system, lessening the risk of high local concentrations.3,4 Minitablets are easy to produce and offer an alternative dosage form for pellets having low porosity, and small batch-to-batch variability.2 On the other hand, the preparation of MUDFs, using minitablets, can become a complex and expensive procedure since the capsule filling is an expeditious process and the minitablet production demands extra care and fine adjustments of the instruments used.3,4
The aim of this research is to design, prepare and evaluate administered orally solid pharmaceutical multi-unit dosage forms of Mesalazine, for targeted release at the intestine area, using capsules filled with matrix minitablets, composed of the active ingredient and the polymeric excipient dextran. These dosage forms were chosen in order to delay the onset of the drug release for a period of time equal to the transit time through the stomach (lag time ≥2 hours) and then gradually release the active substance. The dissolution tests were carried out in various pH media simulating the gastrointestinal track.
Materials and Methods
Materials
The following chemicals were purchased from suppliers and used as received: Mesalazine (5-aminosalicylic acid) and Dextran (from Leuconostoc mesenteroides, average molecular weight 5x106-40x106 ); Magnesium Stearate, and hard gelatin capsules (size 0). All chemicals were of reagent grade and used directly without further purification.
Methods
Preparation of “tablets in capsule” system and powder-filled capsule formulations
In the present study, minitablets were prepared by employing a hydraulic press under constant pressure (4kp) for 1 min and a 7.5mm diameter die. Minitablets consisted of a mixture of the active drug substance (50 mg of Mesalazine) and the excipient (50 mg of dextran). Magnesium stearate (1%) was added as a lubricant. After preparation, 4 minitablets of same consistency where used to fill the size 0 hard gelatin capsules. Also, hard gelatin capsules were filled with a mixture of the drug substance and excipient, each one containing 200 mg of Mesalazine and 200 mg of Dextran.
In vitro drug release studies
The drug release from each capsule was carried out in a USP dissolution paddle at 100 rpm and 37±0.5 °C. All dissolution studies were performed in triplicate. Buffer solutions were used as dissolution media to simulate the conditions in the gastrointestinal tract. For the first 2 h, 400 ml of an HCl solution (0.03M), 0.2 w/v NaOH, pH 1.5 was used (Buffer solution A) to simulate the conditions in the stomach. For the next 3 h, 400 ml of a K2 HPO4 solution (0.2M), pH 9.4 was added to buffer solution A to produce buffer solution B, pH 7.4, in order to simulate the conditions in the small intestine. For the last 7 h, 200 ml H3 PO4 of a solution (0.17M), pΗ 1.5 was added into the vessel to produce buffer solution C, pΗ 6.0, to simulate the conditions in the large intestine. Samples (5 ml) were withdrawn at predetermined time intervals (every 30 mins for the first 120 mins and every 60 mins thereafter), filtered and analyzed using a spectrophotometer. Although Mesalazine shows a maximum absorption at 290 nm, the UV measurements were performed in other wavelengths, as well, in order to increase the method sensitivity. Thus, the measurements of absorbance of the samples within the first two hours (buffer solution: pH 1.5) were performed at λmax = 301 nm. For samples (buffer solution: pH 7.4 and 6.0) the measurements were performed at λmax = 330 nm. As the pH changes towards the neutral and slightly alkaline region, Mesalazine is gradually converted to the forms shown in Figure 1, causing a displacement of the local maximum absorption.
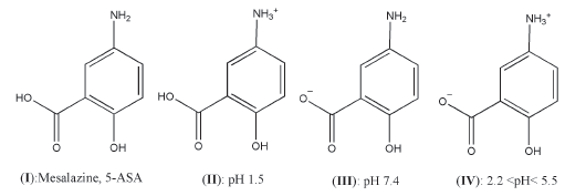 Figure 1. Chemical structures of Mesalazine at various pHs
Figure 1. Chemical structures of Mesalazine at various pHsComparison of the dissolution profiles
In order to compare the dissolution profiles, graphs of % drug release (mean ± standard deviation) vs time were produced and t20%, t50% t90%, and the values of Mean Dissolution Time (MDT) and Dissolution Efficiency (D.E. %) were calculated.
The values of t20%, t50% t90% relate to the time, where the 20%, 50% and 90% of the drug is released. MDT is the value used to characterize the drug release rate from a dosage form and in actual practice, the following Equation 1 is used to derive an estimate of MDT from experimental dissolution data:5

where, W∞ is the asymptote of the dissolved amount of drug and ABC is the area between the cumulative dissolution curve and W∞.
According to Khan (1975),6 D.E. % is a parameter useful for the evaluation of in vitro dissolution and is calculated according to Equation 2:

where, y is the percentage of dissolved product and D.E. the area under the dissolution curve between time points t 1 and t 2 , expressed as a percentage of the curve at maximum dissolution, y100 over the same time period. When a relationship between dissolution and another variable is sought, is considered more realistic to use D.E. %, which takes into account the dissolution profile as a whole. In addition, where a quantitative comparison is required, D.E. % is a more suitable parameter and when limits are set on D.E. % it can be used for quality control in place of the conventional dissolution level.
Results and Discussion
“Tablets in capsule” systems exhibited a different release rate than the powder-filled capsules. The compression of powders to minitablets played an important role in reducing the release rate, t20%, t50%, t90% and MDT, in comparison to powder filled capsules (Figure 2, Table 1). Compression pressure has been identified as a modifier of drug release and the influence of the compression force can be seen in the lag time.7 Specifically, at 120 min in gastric like fluids (pH 1.5), from the “tablet in capsule” system the drug release was 3.74%, while from the powder filled capsule 81.58%. At 300 min, in intestine like fluids (pH 7.4), the drug release from the “tablet in capsule” system reached 15.86%, while from the powder filled capsule 100%. At 720 min the “tablet in capsule” system exhibits a drug release of 100%, showing a modified release formulation across the gastrointestinal track. Hence, this formulation allows Mesalazine to reach the active site (lower part of the large intestine-colon area) almost intact and exert its therapeutic action locally.
 Figure 2. % Release (mean ± SD) of Mesalazine vs time from capsules containing 4 minitablets of mixture with dextran and mixture of powders with dextran and from hard gelatin capsules.
Figure 2. % Release (mean ± SD) of Mesalazine vs time from capsules containing 4 minitablets of mixture with dextran and mixture of powders with dextran and from hard gelatin capsules.Table 1. Types of Mesalazine 200 mg formulations, time at 20%, 50% and 90% of drug release (t20%, t50% and t90%), the Mean Dissolution Time (MDT) and the Dissolution Efficiency (D.E.%).
Figure 2 shows the effect of compression in dextran minitablets on the release rate. Dextran is a polymer, which upon hydration, swells and produces a gelled-layer that acts as a barrier to drug release,8 rendering extended release characteristics to the formulations (values of t20%, t50%, t90% were high, Table 1).
In comparison to the powder filled 200 mg capsules, the “tablet in capsule” system has four 50 mg Mesalazine minitablets inside of one capsule that can be opened. This capsule system permits patients to modify their dosage.
Conclusions
In conclusion, hard gelatin capsules when filled with minitablets lead to a satisfactory modified release profile, compatible with oral administration requirements, due to the thickgelled layer produced by dextran. The data obtained show that dextran-based ‘tablets in capsules’ systems can be used as a model for Mesalazine’s extended release formulations.
References
- Campregher C, Gasche C. Aminosalicylates. Best Practice & Research Clinical Gastroenterology, 2011;25:535-546
- Lopes CM, Lobo JM, Pinto JF, Costa P. Compressed mini-tablets as a biphasic delivery system. Int J Pharm. 2006;323(1-2):93-100.
- Efentakis M, Koutlis A, Vlachou M. Development and evaluation of oral multiple-unit and singleunit hydrophilic controlled-release systems. AAPS PharmSciTech, 2000;1(4):62-70.
- Vlachou M, Siamidi A, Efentakis M. Investigation of a Novel “Tablets in Capsule” Theophylline Formulation System for Modified Release. Pharm Pharmacol Int J. 2017;5(2): 00115
- Rinaki E, Dokoumetzidis A, Macheras P. The mean dissolution time depends on the dose/solubility ratio. Pharm Res. 2003;20(3):406-408.
- Khan KA. The concept of dissolution effi ciency. Communications, J Pharm Pharac. 1975;27:48
- Velasco MV, Ford JL, Rowe P, Rajabi-Siahboomi AR. Influence of drug:hydroxypropylmethylcellulose ratio, drug and polymer particle size and compression force on the release of diclofenac sodium from HPMC tablets. J. Control. Release 1999;57:75 –85
- Casettari L, Bonacucina G, Morris GA, et al. Dextran and its potential use as tablet excipient. Powder Technology 2015;273: 125-132.
Author Biographies
Angeliki Siamidi holds an MPharm degree from the University of Sunderland, UK, finished her MSc in Industrial Pharmacy and PhD in Pharmaceutical Technology at the National and Kapodistrian University of Athens, Greece and is currently a Post-doc researcher at the same University. Her research focuses in modified drug release from solid pharmaceutical dosage forms.
Sofia Konstantinidou is currently an MSc student at UCL, having received an Onassis Foundation scholarship in Clinical Pharmacy, International Practice & Policy. She has participated in a wide range of conferences around the world. She has published four articles in peer reviewed journals and one book chapter.