Executive Summary
In order to advance lifesaving (or disease modifying) therapies to patients, the pressures for expedited development are increasing. Advancements in technology, program needs, and product knowledge inevitably result in the need for comparability assessments driven by late stage process changes. The use of enhanced technologies and risk-based approaches can build robustness in one’s comparability assessments, improve both product and process knowledge, and ensure robust continual product supply to patients. As comparability assessments become increasingly important for evaluating changes during expedited development or in planning for post-approval changes, risk-based approaches and increased agency and industry collaboration are essential enablers for success.
Introduction
The development of biopharmaceutical products is increasingly requiring changes to be introduced late in development or immediately post-launch of the product. Advancements in process or analytical technology, increased clarity on patient requirements and the target product profile (TPP), and/or significant increases in the anticipated patient demand are only a few examples of drivers for change during development. Under typical development timelines, many of these changes can be readily developed, understood, evaluated, and introduced early in the clinical development plan. But to help bring therapies to patients more quickly, many development programs today are expedited. As a result, this speed of development adds time pressure for the development and implementation of process improvements.
Most changes introduced during development are related to the manufacturing process. For complex molecules such as monoclonal antibodies, many manufacturing process changes can affect product quality. To ensure continuity in the clinical program, it is thus important to demonstrate “suitability for use” in early development, or “comparability” during later stages of development between the preand post-change material, and most importantly minimize any risk to patients. As defined in ICH Q5E: Comparability of Biotechnological/ Biological Products Subject to Changes in Their Manufacturing Process,“the goal of the comparability exercise is to ascertain that pre- and post-change drug product is comparable in terms of quality, safety, and efficacy”. As further mentioned in ICH Q5E, comparability does not mean a product is identical post-change but that the pre- and post-change product is “highly similar” and the existing knowledge is sufficient to predict potential impacts to safety or efficacy. In the early clinical context, suitability of use relates to assuring the clinical product meets safety expectations and that non-clinical/clinical data is applicable to the post-change product to ensure continuity in the clinical development and eventual marketing authorization. But it is anticipated that product improvements will be made throughout the course of development, thus the comparability exercise should align with the stage of development. While more comprehensive comparability is typically expected during the later stages of development (pivotal studies) or post-approval. For simplicity, the term comparability will be used more generally in this article in relation to comparing pre- and post-change product.
Under expedited development paradigms, demonstrating comparability with many potential changes can pose significant challenges for both the subject matter experts and regulators. This is particularly true for complex molecules such as monoclonal antibodies, as comparability assessments can take significant time due to the multi-layer analyses often necessary for such complex products (i.e., activity; primary, secondary, and higher order structure; charge variants; post-translational modifications; glycan structure; etc.). Thus, to meet comparability expectations under expedited development timelines, process and product development must be nimble, decisive, and proactive to ensure desired changes can be adequately evaluated and implemented without delaying program timelines and patient access. The philosophy of risk-based comparability paired with regulatory strategy and the increased use of technology can improve the chance of success of demonstrating comparability for complex molecules. Some of these tools include (1) early and transparent regulatory engagement on proposed comparability approaches, (2) the use of protocols in development (designed off the principles defined for post-approval comparability regulatory pathways), and (3) the increased use of analytical and process technologies to expedite product and process understanding. Ensuring these approaches are considered a core component of standard drug development practices when evaluating change during development, can help the company better prepare when change is inevitably introduced into the development plan. This will ensure both process or product improvements, otherwise pushed to the postapproval space due to time constraints, are realized earlier and better able to benefit patients through more robust manufacturing processes leveraging the most advanced technologies.
Risk Based Comparability Approach
For changes made during biopharmaceutical development (i.e. clinical stage), there is an expectation to demonstrate comparability of the preand post-change product. During the course of development product and process knowledge as well as the available analytical toolbox will continue to mature. As a result, the extent and comprehensiveness of the comparability exercise should align appropriately with the stage of development. The application of a risk-based, phase specific comparability approach is recommended to balance the level of risk with the level of molecule understanding, development time, and cost. A risk based comparability approach will help dictate the testing that may or may not be required (analytical, pre-clinical, or clinical testing), commensurate with the associated level of risk to patient safety and the supporting scientific rationale.
Figure 1 provides a hierarchy of the potential testing approach that may be employed, with analytical testing as the first layer of the comparability assessment, followed by non-clinical or clinical studies if analytical comparability cannot be sufficiently determined. This philosophy is in alignment with ICH Q5E, which states “where the relationship between specific quality attributes and safety and efficacy has not been established, and differences between quality attributes of the pre- and post-change product are observed, it might be appropriate to include a combination of quality, nonclinical, and/ or clinical studies”.
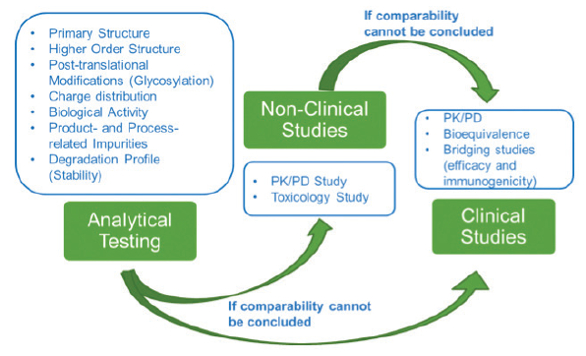 Figure 1. Hierarchy of comparability study approach
Figure 1. Hierarchy of comparability study approachWith the advancement in complex molecule understanding and resulting supportive analytical technologies available today (in particular, advanced biological assays available to probe molecule functionality and extrapolate with confidence to the in vivo context), thorough demonstration of comparability via analytical methods may often be sufficient to mitigate potential risks introduced from a process change. But, it is important to consider if any clinically meaningful differences (in efficacy, safety, immunogenicity) exist between the pre- and post-change material, and to consider the overall potential risk to patients taken in context with the totality of data collected (including any prior knowledge). When taken together, the advancement in analytical technologies allow an increase product understanding, which may reduce the risk of the “unknown” introduced from a process change.
An outline of a general risk based analytical comparability approach is described in Table 1.
Table 1. General risk-based comparability approach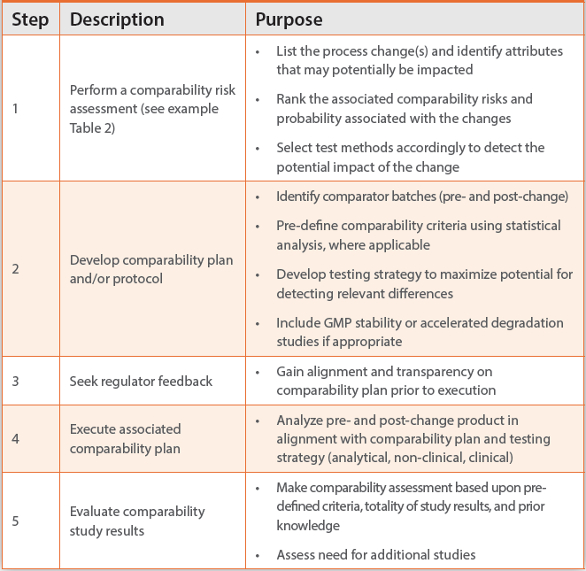
For complex molecules such as monoclonal antibodies, the analytical testing strategy may include, but not be limited to:
- Release tests,
- Elements of full characterization (including primary, secondary, and higher order structure analysis, charge heterogeneity, carbohydrate structure)
- Structure activity relationship studies (to establish the CQAmechanism of action relationship)
- Assessment of product and process impurities
- Stability (DS, DP, or both dependent upon the change(s))
The use of statistical methods, such as equal-tailed tolerance interval (ETTI), can help define the appropriate comparability criteria for the test methods described above. Although challenging in the development space to leverage statistical methods due to limited product and process experience, statistical approaches can help ensure the comparability plan is robust and appropriate for the phase of development, as well as guide development decisions.
Using a pre-determined comparability protocol and risk-based approach can additionally help the company better understand criticality and more clearly justify pre- and post-change differences. This approach can help focus comparability assessments, as well as development time and cost; but should be commensurate with the proposed changes and potential risk to patient safety and product efficacy.
Regulatory Opportunities
To maintain expedited timelines while introducing process change, it is important to consider the partnership between the company and any regulatory agency. Seeking regulatory advice on development approaches can help ensure transparency and early alignment, particularly when it applies to risk-based comparability. Through such engagement, the development team can assess any impact to program timelines, predict the potential for alignment of comparability conclusions with regulators, and ensure the comparability plan is adequately designed prior to devoting significant time and resource. This is particularly important under expedited timelines to ensure process development can proceed with the best technology or approach possible, without risking impact to program timelines or requiring re-development activities. This approach can ensure lifesaving therapies reach patients in need faster.
The core component of the risk-based comparability approach is the comparability protocol. The general concept of comparability protocol pathways is more readily defined in the post-approval space (i.e. CP in US or PACMP in EU), but similar principles can also be applied during development. As defined in the FDA guidance Comparability Protocols for Human drugs and Biologics: Chemistry, Manufacturing, and Controls Information, a comparability protocol is “a comprehensive, prospectively written plan for assessing the effect of a proposed CMC post-approval change(s) on the identity, strength, quality, purity, and potency of a drug product”. The goal of such a protocol is to facilitate subsequent implementation and provide a pro-active approach to help introduce continuous process improvements faster. In the development space, applying aspects of comparability protocols can both expedite the introduction of more commercially viable product and processes, while ensuring minimal impact to program timelines, and while maintaining the integrity of the material used throughout clinical studies.
Table 2. Example comparability risk assessment for common process changes during development
Engaging regulators with a well thought out comparability protocol can help foster collaboration. Although agreement on the conclusions of the comparability assessment may not be possible based on the protocol alone without the study data, proactive communication will support alignment on key study expectations. When expedited timelines are the result of accelerated pathways (i.e. SAKIGAKE, PRIME, Breakthrough Therapy, etc.), the opportunity for such advice may be more readily available and should be taken advantage of.
Technology/Strategy in Expedited Development
Demonstrating comparability during development can often be challenging due to limited product and process knowledge. Knowledge Management thus becomes a critical component for managing change(s) and assessing comparability. The use of technology can help accelerate product and process understanding during development, and can additionally provide monitoring capabilities to ensure adequate control and process consistency is maintained post-change implementation. The use of peptide map based multi-attribute methods, process modeling, and short accelerated stability studies are a few examples of technologies and strategies that can help manage change during development in conjunction with continuing to build product and process knowledge. Table 3 summarizes the potential use of each technology, what benefit each may provide during expedited development, and the challenges that may be faced. Each application can provide complementary value when coupled with standard comparability approaches by reducing risk through increased oversight post-change implementation and increased product and process understanding gained during development.
Table 3. Technology to support comparability assessment during development
The multi-attribute method can be used to rapidly evaluate proposed process changes and aid in process development decisions, for example during clone selection or to support DoE based experiments with many potential experimental conditions. Process modeling can be used to statistically evaluate how consistent and comparable the pre-and post-change manufacturing processes perform in comparison to “golden” or target manufacturing performance. This can help assess at scale comparisons and could be used to continuously monitor performance once the process change is implemented. Short accelerated stability studies can be supportive of existing GMP stability studies, and are amenable to quickly detecting any new degradation species. When coupled with the risk based comparability approach, short accelerated stability studies can help complement traditional real-time stability at the time of implementing the change(s).
These three examples are only a subset of the many potential tools and emerging technologies that can expedite knowledge gathering during process development and/or mitigate risks post-implementation. It is important to note the utility of such technologies or strategies is dependent upon having historical data generated using each approach. Thus, it is important to either integrate these technologies/ strategies early in development to build a foundational data set, or pro-actively save representative retain samples in anticipation of future technology/strategy introduction.
Conclusions
Bringing life-changing therapies to patients suffering from unmet medical needs quickly is often both an ethical and business priority in biopharmaceutical development. As a result, development timelines are often expedited; challenging both subject matter experts and regulatory professionals who want to ensure the most robust product and process can be put in place prior to commercialization. Often beneficial process changes get pushed to the post-approval stage due to limited product and process knowledge during development, limited time for assessment, and uncertainty in the potential success of any expected comparability assessments. By (1) incorporating proactive risk-based comparability approaches into standard development practices, (2) increasing regulatory engagement for transparency and alignment on strategy, and (3) leveraging technology to increase product and process knowledge, process and product improvements can be realized faster, ultimately benefiting patients.