The comprehensive nature of drug development, combined with the demands of multiple stakeholders, requires drug developers to manage both resources and risk if they are to efficiently bring new products to market. The sequential, interdependent nature of the drug development process means that each decision can have a major impact on future development.
Not carrying out a thorough assessment on a drug molecule’s solid-state properties, or not considering a solubility-enhancing technology during pre-clinical development, may increase the overall time and cost of a project when sub-optimal bioperformance properties are seen later in phase 1 and 2 clinical trials.
The new generation of small molecule drugs have inflated molecular weights and lipophilicity when compared to the drugs discovered in the 1990s and earlier. These new, complex molecules not only pose biopharmaceutical challenges such as poor solubility and bioavailability, but can also bring handling and manufacturing challenges such as potent handling requirements. Less than 30% of new chemical entities in drug development pipelines are readily bioavailable when administered orally,1 so when orally dosed in patients, these drugs are less likely to attain sufficient exposure (efficacy), and are likely to have higher variability in exposure (safety issues). To develop a drug with optimal bioperformance, drug formulators must understand and optimize molecular characteristics and possibly employ solubility-enhancing technologies.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
Approaches to Formulation Strategy
A molecule’s ability to solubilize and permeate across the gut wall are the main determinants of its oral bioavailability and form the basis of both the Biopharmaceutics Classification System (BCS) and the Developability Classification System (DCS).2,3 The DCS (shown in Figure 1) modifies the BCS to be used as a formulation tool, in comparison to being a regulatory tool for biowaivers. The DCS examines the total dose, instead of the highest dose strength, when assessing a drug’s solubility, and shifts the border between low and high solubility from 250 mL to 500 mL Class II in the DCS is divided according to molecules whose absorption is dissolution rate-limited (DCS IIa) and solubility-limited (DCS IIb).
To obtain the best predictions from the BCS and DCS classification tools, accurate measurements of permeability and solubility are needed for the final polymorph and salt form of the drug candidate. This further emphasizes the need for thorough preformulation data prior to choosing a formulation strategy. However, even with ideal data input, the DCS is not a steadfast rule, and does not minimize or eliminate the need for formulation screening, as there may be cases where DCS class IIa drugs require solubility enhancing technology, such as a lipid based drug delivery system (LBDDS), to improve absorption.
In addition to the biopharmaceutics considerations, if a drug is to stand the best possible chance of proceeding through development, scale-up and commercialization efficiently, then formulators must consider the quantity of active ingredient available for testing during early pre-clinical stages, both short and long-term manufacturing considerations, future scale-up strategies, and patient considerations such as dosing regimen and acceptability for the specific patient group.
Choosing Formulation Strategies
To demonstrate the many ways drugs can be optimized and developed, below are four potential scenarios, each following the progression of theoretical molecules, and showing how developmental scientists are influenced by the drugs’ physicochemical and biopharmaceutical properties to determine their formulation strategy and dosage form development.
Scenario 1
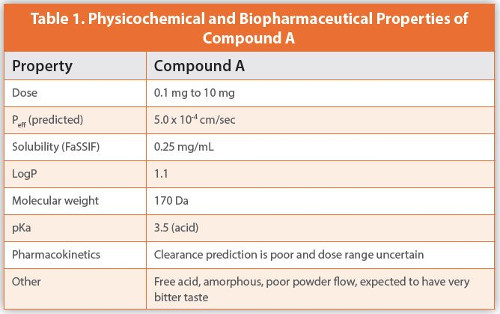
Compound A is a new chemical entity with the properties summarized in Table 1. These include an estimated apparent human jejunal permeability (Peff) of 5.0 x 10-4 cm/sec, and the solubility in fasted state simulating intestinal fluid (FaSSIF) is determined to be 0.25 mg/mL.
Based on the given physicochemical data, compound A is classified as Class I in the DCS (at highest dose of 10 mg) due to its high solubility and high permeability and demonstrates a relatively easy molecule to develop. It is a free acid with a pKa of 3.5, so it will be present in an ionized form in the intestines leading to less absorption. However, the majority of the drug will be non-ionized in the stomach, allowing greater absorption there.
The estimated clinical dose indicates that the Active Pharmaceutical Ingredient (API) could potentially be a highly potent molecule that may require special containment during manufacture and analytical testing, and with the minimum dose being 0.1 mg, this can lead to potential dose uniformity issues.
The potentially wide dose range, and the API powder having poor flow properties, rules out the approach of dosing the API neat into gelatin or hydroxypropylmethyl cellulose (HPMC) capsules. This is because the dose needs to be within the technical dosing limit of the equipment (0.1 mg to 200 mg) for filling the powder into a capsule, so the remaining option, which is the simplest, most cost effective and fastest route to clinic, is filling a solution of the drug in a bottle.
To overcome the potential bitter taste, the addition of a flavor such as peppermint oil could be utilized as a taste masker. If the amorphous form is stable, it is most likely that the drug solution in a bottle will provide a stable formulation. However, a polymorph screening is highly recommended to confirm the stability of the amorphous form. Samples should be tested for physical stability by powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC) to confirm that the drug in an amorphous state is stable against crystallization. Because of the very low dose, content uniformity of the drug in solution is of a lesser concern, however, analytical testing and specifications will be challenging at the lowest strength of the drug. This compound’s uncertainty around clearance could have a potential impact on the clinical dose range, and this would require further investigation.
Scenario 2
Compound B requires a plan for formulation, pre-clinical toxicology and phase 1 clinical studies. As well as the properties of the compound (Table 2), screening may be limited by availability of the API and by budget constraints.
Salt formation is not appropriate for this scenario as the compound does not contain an ionizable functional group. Since it has two crystal forms, it is critical to know which one of the two forms is thermodynamically stable in its current polymorphic form and its anticipated stability during formulation of the drug product or storage. Polymorphic form also has an impact on a compound’s physical characteristics that can affect bioavailability. Therefore, a polymorph screen is highly recommended, followed by physicochemical characterization using DSC, PXRD, Fourier-transform infrared spectroscopy (FTIR), dissolution/solubility, and stability testing to determine the best polymorphic form of compound B.
By plotting Peff against solubility/dose ratio, compound B falls into Class IIa of the DCS; which means that the dissolution rate is the limiting factor for drug absorption. Complete solubility is defined by the compound being freely able to disperse and dissolve. Therefore, to enhance its oral bioavailability, reducing the compound’s particle size is recommended.
Micronization or comicronization will increase both the surface area and wettability of the drug particles, and result in an enhanced dissolution rate. The DCS includes a proposed equation to calculate a target particle size (D90), which represents the diameter below which 90% of the sample lies.3 In this case, the target particle size (D90) is 77 μm. The DCS further defines a solubility-limited absorbable dose (SLAD) of 250 mg. As the intended dose is below the SLAD value, it is predicted that absorption of the complete dose will be sufficient, and will not be limited by its solubility.
Particle reduction is a simple, lower-cost approach that can be applied to projects with limited budget. Depending on the final properties of the micronized compound, it can be directly encapsulated in hard gelatin capsule, or further formulated in a conventional powder blend/granule tablet. Screening of the micronized prototype formulation within preclinical and stability studies should be conducted ahead of the selection of an optimized formulation for phase 1 clinical supply manufacturing.
Scenario 3
Compound C requires a strategy for the formulation development, pre-clinical toxicology and first-in-human studies. Its properties are listed in Table 3, including its low solubility in FaSSIF, leading to low bioavailability. In vitro cell permeability studies and microsomal studies showed that the molecule is a P-glycoprotein (P-gp) substrate and 25% of the drug is metabolized by liver enzymes, respectively.
Compound C is classified as a DCS IIb compound with a SLAD of 100 mg. As the intended dose is above the SLAD, it suggests that the drug’s limited intrinsic solubility is the limiting factor for absorption, therefore bioavailability enhancement through formulation is warranted.
Since compound C does not have any ionizable functional groups, salt formation is not feasible. Typical bioavailability enhancing technologies for DCS IIb compounds include amorphous dispersions and lipid-based drug delivery systems (LBDDS). As the drug is already in an amorphous state, LBDDS appears to be the best option. As listed in Table 3, compound C is a P-gp substrate. Certain LBDDS excipients such as vitamin E TPGS, polyethylene glycol, Labrasol® and Polysorbate 80 may have P-gp inhibiting properties and may be helpful in mitigating losses due to first pass metabolism.4
The next step in the development of compound C would be to determine which excipients or combination of excipients are best suited for a robust formulation strategy. A comprehensive preformulation strategy that includes solubility screening of the molecule in lipid excipients, biorelevant media, and lipid digestion products must be performed to determine useful excipients. In addition, the stability of the molecule in the excipients and any risk of precipitation must also be evaluated. After stable prototype lipid formulations are identified, they can be encapsulated into soft gelatin capsules which are ideal for most lipid-based oral formulations. These prototypes must commence stability studies, and be dosed to animals in a pre-clinical bioavailability study, prior to selection of a lead formulation for first in-human clinical trial manufacture.
Scenario 4
Compound D is a DCS Class IIb molecule with a SLAD of 67.5 mg. In addition, the molecule is a free base with a pKa of 4.5, suggesting higher variability in absorption due to variability in stomach pH of oncology patients, who are often prescribed acid reducing agents. The molecule has the potential to be granted “breakthrough therapy” and “fast track” designations by the U.S. Food and Drug Administration (FDA) if successful in phase 1, and if it is, then the program could rapidly accelerate to later phases. As such, early development to solve any solubility challenges, and formulation design should allow for fast and efficient scale up into late phase and commercial products. The molecule has a narrow therapeutic window, meaning a formulation approach that gives flexibility to change dosage strengths in the future must be considered.
As discussed previously, polymorphic forms can affect bioavailability, so a thorough polymorph screen must be undertaken to determine whether the most stable polymorphic form for compound D can be manufactured. Since the intended dose is significantly higher than the SLAD, solubility enhancement techniques such as LBDDS and amorphous dispersions (spray drying and hot melt extrusion) need to be explored.
With this information, a parallel formulation screen can be undertaken, to investigate solubility in lipid excipients, and miscibility in polymers used in amorphous solid dispersions. These studies are performed to assess which lipids, excipients or polymers and drug loadings have the best potential.
These materials must be tested in vitro using a kinetic dissolution model to screen amongst technologies for comparison to crystalline free base API. Simultaneously, the best performing formulations must be tested in vivo in a pre-clinical bioavailability study.
This allows the best performing technologies to be identified and, subject to excipient compatibility studies, considered for first in human clinical trial manufacture.
If a salt form offers higher solubility without any precipitation in stomach pH, the salt form of the drug can be formulated into a simple blend/granule and can be filled into a capsule or tableted. However, the moisture content and general handling of the formulation must be considered. In addition, due to the high expected therapeutic dose, the extra mass of the salt in dosage form should be considered.
If the drug has sufficient solubility in lipid excipients, LBDDS formulation in soft gelatin capsules is a promising pathway for relatively straightforward and rapid scale-up to clinical and commercial supply. In addition, lipid based formulations have strong potential to mitigate the positive food effect. This is an important consideration because it would not only improve solubility and absorption, but also reduce pharmacokinetic variability and the potential for overdosing.
If hot melt extrusion provides the desired solubility, the material can be filled directly into a capsule or tableted with the aid of minimal excipients. However, API resources must be considered for this technology, since it uses a substantial amount of API during scale-up optimization. If spray drying proves to be preferable, the material can be roller compacted with the help of additional excipients and tableted. However, thorough moisture controls must be established during general handling of the dispersion.
Conclusions
There are numerous articles summarizing the benefits of bioavailability enhancing technologies, however, in this article, the intention was to illustrate the application of the formulation concepts through the journeys of four theoretical molecules.
The scenarios aimed to demonstrate that a comprehensive parallel evaluation of formulations can prove to be beneficial to overcome the challenges posed by the physicochemical and biopharmaceutical properties of drug molecules. The DCS can provide a starting point and offer guidance as to the reasons for poor drug absorption (such as low intrinsic solubility or low dissolution rate) and potential solutions in terms of enhancing technologies.
Each molecule offers its own unique challenges, but by using the DCS, and having access to a number of formulation and delivery technologies, a formulator can evaluate a drug’s properties, such as excipient miscibility and pharmacokinetic considerations, to plan the best approach for a drug undergoing preclinical toxicology and phase 1 studies.
References
- Bayliss MK, Butler J, Feldman PL, Green DV, Leeson PD, Palovich MR, et al. Quality guidelines for oral drug candidates: dose, solubility and lipophilicity. Drug Discov Today. 2016;21(10):1719-27.
- Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413-20.
- Butler JM, Dressman JB. The developability classification system: application of biopharmaceutics concepts to formulation development. J Pharm Sci. 2010;99(12):4940-54.
- Wang SW, Monagle J, McNulty C, Putnam D, Chen H. Determination of P-glycoprotein inhibition by excipients and their combinations using an integrated high-throughput process. J Pharm Sci. 2004;93(11):2755-67.
Labrasol® is a registered trademark of Gattefossé