Translational Sciences, Seattle Genetics
You wake up in the morning. Amazon Alexa tells you that today is mostly cloudy but will rain at noon. She also reminds you to take your daily medications and to wish your sister happy birthday. You scroll through your Facebook feed and an ad appears that tells you a restaurant is now serving your favorite gingerbread pancakes. You start your car engine, and as you being to drive to work, Google Maps tells you an alternative route should save you 20 minutes. You arrive at your office and your boss asks you to shop for a 3D printer for a “delicious” project, printing real cakes. You find a good-ranking model at Amazon, which also automatically recommends all of the cake-printing supplies you need. This type of thing is happening every day to most of us. It is artificial intelligence (AI), or at least, the start of an AI era. Alexa, or Apple’s Siri, uses speech recognition designed to understand a user’s request and return a response. Facebook’s AI knows who you are from photos and videos you’ve uploaded and conversations with friends, and decides which news feeds, friend recommendations, and ads you receive. By definition, artificial intelligence is a computer-aided modeling process that solves cognitive problems such as learning and pattern recognition, which are aspects of human intelligence.
Artificial intelligence comes from sophisticated computer model based “machine learning” and “deep learning”. Microsoft and IBM provide AI infrastructure using modeling tools. Farmers are able to develop intelligent planting, irrigating and harvesting systems to increase yields to feed the expanding population of our planet. Algorithms and data, often “big data” (defined as data with large volume, high velocity and complex variety), are the core of modeling and learning. Wall Street quants build complex mathematical models to predict stock prices for maximized profits and minimized risks. J. P. Morgan develops “trading bots” to execute trades, which model and make decisions using your data from the moment you type a buy/sell price from your cell phone. Big data feed machine-learning algorithms to acquire artificial intelligence.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
In drug discovery and development, modeling and simulation have also been explored and gradually applied in many areas recently. The Human Genome Project was successfully completed in 2003, which has provided an enormous amount of genetic data for deep learning about a disease or target for drug discovery. Over 15 years later, drug discovery has made significant improvements because of the deeper understanding of drug targets. However, many diseases, especially most types of cancer, still have no cure. At best, we can maintain the disease condition to extend life for months or even years. The human body is astronomically complex. Diseases, if not genetic, are generally either life-style-related such as cardiovascular diseases, diabetes and obesity, or age-related such as Alzheimer’s and rheumatoid arthritis. Cancer can be both. Many signal pathways are involved with cancer; these combinations make it extremely difficult to find a cure. The role of the immune system makes it even more complicated. Modeling aspects of the human body such as the immune system requires more advanced physiology-based modeling and AI.
Drug Metabolism
Alexa reminded you this morning to take a 10 mg atorvastatin tablet for your slightly elevated blood cholesterol level. What happened to the tablet in your body after you swallowed it is the science of drug metabolism, or more accurately, drug metabolism and pharmacokinetics (DMPK). DMPK studies the absorption, distribution, metabolism and excretion (ADME) of a drug. The birth of drug metabolism as a distinct branch of science began in the 1960s, particularly after the discovery of the role of cytochrome P450s in metabolism. Atorvastatin is metabolized by a specific P450 called CYP3A4 into several more polar metabolites, and they leave the body mainly by biliary excretion with a tiny portion in urine. While the drug is available in other dosage levels such as 20, 40 or 80 mg tablets, you may only need to take 10 mg once daily, as Alexa reminds you every morning. All these details are related to drug metabolism, which determines the efficacy and safety of atorvastatin.
Alexa also reminded you this morning not to drink grapefruit juice with atorvastatin. This is a part of drug metabolism as well, or drug-drug interactions (DDI). A special ingredient in grapefruit called bergamottin inactivates CYP3A4 via formation of covalently bound electrophilic metabolites. Thus, the net effect of eating two grapefruits with a 10 mg tablet may actually be like taking a 20 mg tablet, a possible overdose. A crazier example of overdosing by drug-drug interaction is combining cocaine and alcohol. The combination is popular among drug users for more intense highs, which also causes increased incidence of sudden death. When cocaine and alcohol coexist in the blood, drug-metabolizing enzymes catalyze the formation of a toxic metabolite called cocaethylene.
Drug metabolism is a tiny but exciting area in the pharmaceutical industry. For the last 10 years, drug metabolism assays and animal studies have mainly been outsourced. Actually, small companies are mostly virtual in DMPK. DMPK scientists that remain these days in pharma and biotech mainly monitor outsourced studies and integrate data for project teams. Data integration, which includes data analysis, modeling and prediction, is a key process to aide in decision making. Creativity is required more than ever these days for drug metabolism. Modeling, simulation, high-throughput data analysis and “smarter” data integration are key to keep drug metabolism as a unique core function in today’s drug discovery.
Modeling in Drug Metabolism
Thousands of years ago, people already knew how to use models to improve everyday life. For example, Egyptian sundials, Hindu-Arabic numerals, Babylonian sexagesimal numerals, and Chinese abacus are prototype modeling systems. People use models to explain phenomena and/or make predictions for communication and decision-making. This applies to modeling in drug metabolism as well. Several categories of modeling in drug metabolism are ADME data modeling, pharmacokinetic (PK) modeling, data mining, structure-based modeling, and physiologically-based pharmacokinetic (PBPK) modeling.
Data modeling. The simplest modeling in drug metabolism is to model enzyme kinetics with linear or nonlinear equations. Examples are Michalis-Mention kinetics of P450-catalyzed reactions and 4-parameter logistic modeling of P450 inhibition/induction. Major CROs and pharmaceutical companies automate and customize computer applications to handle routine data analysis and modeling on a large scale. With the development of computer algorithms, thousands of data analysis procedures can be accomplished in seconds, with high-quality tables and figures generated instantly. A SAS-based application called AIR Binder (ADME Instant Report Binder) was recently developed that automatically performs data analysis/modeling, dynamic visualization and reporting of preclinical and clinical ADME assay data.1,2 Based on our experience, productivity was significantly enhanced with the use of AIR Binder, which is attributed to its use of object-oriented programming concepts to organize SAS macros, together with SAS’ powerful statistical analysis capacity and ODS graphics.
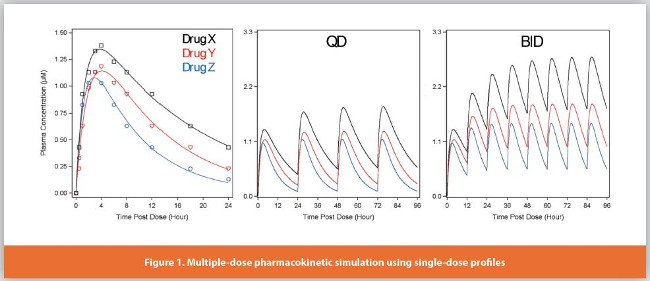
PK modeling. Pharmacokinetic modeling with non-compartment analysis (NCA), one compartment, or multiple compartments is routinely used in drug metabolism to explain time-course drug concentration data for both small molecules and biologics. These models are derived from exponential functions and differential equations. Multiple-dose modeling simulated by using single-dose data can help the design of efficacy and safety studies, which is a “smarter” quantitative approach for drug metabolism. On the platform of AIR Binder, we built a PK simulator called ψ (PSI, Pharmacokinetic Simulation Interface), which models and simulates the user-defined dose and dosing schedules instantly (Figure 1). PK/PD modeling has been widely implemented to correlate the exposure of a drug with its efficacy/safety - the pharmacodynamics (PD) properties of a drug. In addition, the starting dose for first-in-human clinical trials have been modeled with the NOAEL-based approach (no observed adverse effect levels), or more recently, the MABEL-based approach (minimal anticipated biological effect level), which is another example of PK/PD modeling.
Data mining. When you stop your car at an empty traffic light after midnight, have you wondered why it takes only a couple seconds to turn green in big cities like Seattle but may take two minutes in a small Wisconsin town? It is likely attributed to IoT, or internet of things. IoT is a system of chips or devices connected to the Internet that can send and receive data without requiring human interaction. An adaptive IoT transportation management system (installed in most big cities) enables traffic lights to adapt to changing road and weather conditions. The data processing performed with IoT is the simplest type of data mining. High-throughput screening has given large pharma a gigantic database from which they can mine useful information for metabolism-derived drug design and lead optimization. Large datasets from metabolic stability, passive/active permeability, and P450 inhibition assays have been routinely mined to build QSAR (Quantitative Structure-Activity Relationship) models. For example, from the database of P450 inhibition by pyridine-containing compounds, QSAR models have been built from the 2-, 3-, or 4-pyridyl inhibition patterns on CYP1A2, 2C9, 2D6, and 3A4 enzymes.3
Instead of mining quantitative data, another application of data mining is on patterned qualitative data, such as those from mass spectrometry. Mass spectrometer has a long history. In the Manhattan Project, it was used to separate isotopes of uranium. In 1976, NASA sent mass spectrometers to Mars to analyze Martian atmosphere and soil. Since John Fenn and Koichi Tanaka’s development of soft desorption ionization methods in 1980s (Nobel Prize in chemistry in 2002), mass spectrometry has gradually become the core of drug metabolism research for bioanalysis and metabolite identification. Metabolite identification and characterization using mass spectrometry is a process of pattern recognition. Molecular ions are selected by the mass spectrometer, with high-energy collision breaking the molecule into fragments. The resulting fragmentation patterns are fingerprints for structural elucidation, which traditionally need highly trained personnel to manually explore. The patterns of small molecules are relatively simple, but today’s drug discovery has diverse chemical moieties such as peptides, oligonucleotides, and antibody-drug conjugates, which make metabolite characterization more complicated than ever. Recently, we put together a Python-based application, LEO (Linearization, Extension and Oxidation), to generate solutions for relatively challenging molecules.4,5 The algorithm was built to search theoretical metabolic pathways: cleavage reactions (“linearization”), conjugation reactions (“extension”), and oxidation-reduction reactions (“oxidation”). Due to the difficulties of manually determining all possible combinations of these reactions, an advantage of computer modeling is that it improves efficiency and shortens the turnaround time of metabolite identification.
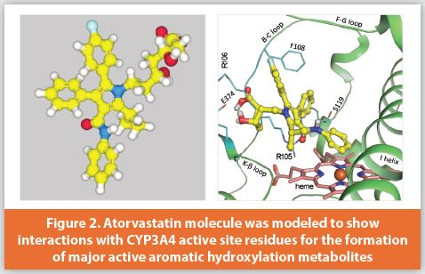
Structure-based modeling. X-Ray crystal structures of the drug target proteins have been widely used for drug design and discovery, which is a structure-based rational drug design approach. X-ray crystallography is a century old. The father and son team Sir William Henry Bragg and William Lawrence Bragg won the Nobel Prize in physics in 1915 for their work in X-ray crystal structures. Rosalind Franklin’s X-ray diffraction image (so-called “Photo 51”) was crucial data that led to the development of a double helix DNA model by James Watson and Francis Crick in 1953 (and their Nobel Prize in physiology/medicine in 1962). Advancements have been made over the last 20 years to crystalize drug-metabolizing enzymes. Currently, all major human P450 crystal structures have been determined at a very good resolution, often co-crystalized with probe substrates or inhibitors. These crystal structures, along with structure-based modeling such as molecule docking, molecular dynamics, and the hybrid quantum mechanics/molecular mechanics (QM/MM) modeling, have been used in metabolism-derived drug design to solve metabolism-related problems such as regioselectivity, stereoselectivity, reactive metabolite formation, metabolic switch, mechanism-based inactivation, and drug-drug interactions.6-12
Most non-P450 drug metabolizing enzymes have also been crystalized and modeled for drug design and lead optimization. Aldehyde oxidase (AO) catalyzes the metabolism of various azaheterocycle-containing drug molecules. A series of structural analogs of zoniporide were modeled for metabolic clearance prediction by altering their molecular interactions with the active site residues of AO.13 Carboxylesterase is the enzyme responsible for the metabolism of cocaine via a hydrolysis reaction. Carboxylesterase-catalyzed reactions were modeled to design efficient ester prodrugs.14 Several major classes of glutathione S-transferases (GSTs) were also crystalized; such enzymes catalyze glutathione conjugation of reactive metabolites or covalent-binding drugs such as acalabrutinib, a novel BTK inhibitor for the treatment of mantle cell lymphoma.15 To facilitate the application of various third-party modeling tools, we developed a Python-based application, MARS (Molecular Modeling and Rendering Solver), to automate the structure-based modeling process for improved efficiency, interpretation and visualization of modeling results (Figure 2).
PBPK modeling. Thirty or forty years ago, and perhaps still today in some countries/places, the term “pediatric drug” did not exist. If a 5-year old is getting sick, the parent may just give him/her a half or a third portion of the adult dosage. Although it is still challenging to get clinical trials for certain pediatric population today, PBPK modeling helps predict the right dose level and schedule based on general population PK and the unique physiological parameters of a child. PBPK modeling applies physiologically realistic compartmental structure by combining sets of differential equations to simulate pharmacokinetic profiles in a special population and/or situation, such as pediatrics, drug-drug interactions, hepatic/renal impairment, food/juice effect (such as grapefruit juice), and pharmacogenetics.
In addition to PK parameters (such as dose, route and frequency of administration) and physicochemical properties of a drug molecule (related to permeability, partitioning to tissues, plasma protein binding, etc.), physiological parameters such as age, weight, height, and genetic features are also integrated in PBPK models. These physiological parameters can be very difficult to collect and complicated to generalize for a model, which is why intelligent algorithms are required. For the last few years, regulatory agencies such as the FDA and EMA have encouraged drug companies to simulate untested scenarios based on key clinical data. For example, when evaluating the DDI potential of a drug candidate as a “victim” (substrate of an enzyme/transporter), a strong inhibitor will normally be selected in clinical trials. Weak inhibitor scenarios can be simulated with PBPK models that have been refined with derived strong inhibitor clinical data, which is a useful process to address information gaps, enhance benefit/risk assessment and properly inform prescription drug labeling.
Future Modeling in Drug Metabolism
There are no perfect models. We are using models to improve communication, help decision making, and ultimately to achieve artificial intelligence, hopefully. For modeling in drug metabolism, sometimes we have too many assumptions and parameters for a model but not enough data collected, which makes the model less robust or “over-fit”. Many factors need to be considered for model optimization, such as covariance of parameters for statistic models, appropriate descriptors to choose, or time-scale dynamic change of protein 3-D structures. The human body is a dynamic system that can adapt to many changing things, good or bad. The lack of adequate and reliable system data, which can be attributed in part to changing physiological parameters with age and disease status, presents major challenges for PBPK modeling. IoT may help in the future. For example, an ingestible pill-sized sensor may be used, which gets activated upon swallowing and starts gathering data from patients. Another factor that presents challenges for modeling is the placebo effect. Love, hope, care, and sprit all can change conditions of the human body; sometimes, the placebo effect is huge.
With the start of an era of AI, big data mining, and deep learning, the gap to realistic drug metabolism models is narrowing. “Smarter” modeling in drug metabolism and discovery is on the near horizon. Every industry is working hard now on modeling and developing artificial intelligence. Self-driving cars are in final testing for production. Ford’s Argo and GM’s Cruise are making progress. People are even talking about Google’s flying cars. SpaceX has identified its first space tourist, and Blue Origin expects to begin selling tickets for space travel this year. Elon Musk says he is on track to launch people to Mars within six years. This is an era of great technology advancement. In the not too distant future, smart modeling and artificial intelligence will be fully integrated into drug discovery and development, and also into our tiny but exciting drug metabolism field.
References
- Sun H, Cardinal K, Voorman R. AIR Binder 2.0: a dynamic visualization, data analysis and reporting SAS application for preclinical and clinical ADME assays, pharmacokinetics, metabolite profiling and identification. MWSUG 2017: PH04; 1-26.
- Sun H, Cardinal K, Kirby C, Voorman R. AIR Binder: an automatic reporting and data analysis SAS application for cytochrome P450 inhibition assay to investigate DDI. PharmaSUG 2017: PO09; 1-8.
- Sun H, Scott DO. Using crystal structures of drug metabolizing enzymes in mechanismbased modeling for drug design. In: Renaud J-P, ed. Structural Biology in Drug Discovery: Wiley, 2019 (in press).
- Cardinal K, Sun H. Metabolite identification and characterization by mining mass spectrometry data with SAS and Python. PharmaSUG 2018: AD34; 1-5.
- Sun H, Cardinal K. Mining metabolites of peptides and antibody-drug conjugates from mass spectrometry data using SAS and Python. SASGF 2018: 2510; 1-6.
- Sun H, Bessire AJ, Vaz A. Dirlotapide as a model substrate to refine structure-based drug design strategies on CYP3A4-catalyzed metabolism. Bioorg Med Chem Lett 2012;22:371-376.
- Sharma R, Sun H, Piotrowski DW, et al. Metabolism, excretion, and pharmacokinetics of ((3,3-difluoropyrrolidin-1-yl)((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrol idin-2-yl)methanone, a dipeptidyl peptidase inhibitor, in rat, dog and human. Drug Metab Dispos 2012;40:2143-2161.
- Miao Z, Sun H, Liras J, Prakash C. Excretion, metabolism, and pharmacokinetics of 1-(8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl)-4-(ethylamino)piperidine-4-carboxamide, a selective cannabinoid receptor antagonist, in healthy male volunteers. Drug Metab Dispos 2012;40:568-578.
- Sun H, Scott DO. Metabolism of 4-aminopiperidine drugs by cytochrome P450s: molecular and quantum mechanical insights into drug design. ACS Med Chem Lett 2011;2:638-643.
- Sun H, Scott DO. Structure-based drug metabolism predictions for drug design. Chem Biol Drug Des 2010;75:3-17.
- Sun H, Sharma R, Bauman J, et al. Differences in CYP3A4 catalyzed bioactivation of 5-aminooxindole and 5-aminobenzsultam scaffolds in proline-rich tyrosine kinase 2 (PYK2) inhibitors: retrospective analysis by CYP3A4 molecular docking, quantum chemical calculations and glutathione adduct detection using linear ion trap/orbitrap mass spectrometry. Bioorg Med Chem Lett 2009;19:3177-3182.
- Sun H, Piotrowski DW, Orr STM, et al. Deuterium isotope effects in drug pharmacokinetics II: Substrate-dependence of the reaction mechanism influences outcome for cytochrome P450 cleared drugs. PLoS One 2018;13:e0206279.
- Dalvie D, Sun H, Xiang C, Hu Q, Jiang Y, Kang P. Effect of structural variation on aldehyde oxidase-catalyzed oxidation of zoniporide. Drug Metab Dispos 2012;40:1575-1587.
- Sun H. Capture hydrolysis signals in the microsomal stability assay: molecular mechanisms of the alkyl ester drug and prodrug metabolism. Bioorg Med Chem Lett 2012;22:989-995.
- Podoll T, Pearson PG, Evarts J, et al. Bioavailability, biotransformation, and excretion of the covalent BTK inhibitor acalabrutinib in rats, dogs, and humans. Drug Metab Dispos 2019; 47:155-163