Abstract
Expedited development of biopharmaceuticals requires a focused, consistent and efficient analytical development strategy. For commonly monitored critical quality attributes, robust platform methods can be implemented and used for a defined class of molecules thus minimizing the need for additional development. For new compounds, or when new attributes need to be monitored, additional method development will be required but this process can be accelerated by adopting a focused approach consisting of four stages with well-defined deliverables (method definition, technology selection, method development/optimization, and method performance evaluation).
Once the method requirements are defined and the initial method established, the key method parameters with the biggest impact on the method performance are identified through systematic assessments of the different method steps. Development activities are then prioritized to minimize the time needed to create an intrinsically robust method which meets the pre-established method requirements and is adequately controlled to prevent drift during routine use. Job aids, method assessment templates, and standardized method validation strategies can be implemented to ensure the development approach is consistent within an organization and to reduce method development time and cost.
Introduction
The availability of reliable analytical methods to monitor the quality attributes of a new biological entity (NBE) is critical for guiding process and product development activities throughout the different product development stages. Analytical methods are also required to monitor the drug substance and drug product to assure that applicable standards of identity, strength, quality, and purity are met. During early-stage development the analytical results are used to support candidate selection, pre-formulation screening, and to confirm that the materials created using the initial early-stage drug substance (DS) and drug product (DP) manufacturing process are suitable to support the non-clinical safety and first in human (FIH) studies. In addition, data from analytical comparability studies are used throughout a program to demonstrate that changes made to the drug substance and drug product manufacturing process do not adversely impact the critical quality attributes associated with efficacy and safety.
Over the years, multiple regulatory approval pathways have been introduced for rapid commercialization of new medicinal products such as the breakthrough therapy designation in the United States, PRIME in the European Union, and Sakigake designation in Japan.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
This has created an additional need to reduce the time required to establish suitable methods to support process and product development activities and create the analytical control strategy. In this paper, we will focus on approaches which can be used to expedite the development of robust GMP methods to facilitate rapid development of NBEs.
Role of Analytical Methods in The Product Control Strategy
To expedite drug development it is necessary to have an understanding of the relevant quality attributes of a new entity to ensure the analytical development activities are consistently focused on the key methods needed to monitor these attributes. The critical quality attributes (CQAs) are derived from the quality target product profile (QTPP) and take into consideration structure function information available for the NBE or class of NBEs. For common entities such as monoclonal antibodies the CQA assessment can be based on platform knowledge as well as literature information. Once identified, the CQAs are used to drive the process development activities to ensure robust unit operations are established which result in a consistent and well-controlled product. A thorough understanding of the impact of the different unit operations on the CQAs forms the basis for establishing the overall control strategy through a combination of process control, analytical control, site control, raw material control, and contaminant control measures. A schematic overview of the relationship between product understanding, process understanding and the control strategy is provided in Figure 1.
The Analytical Method Development Process
The goal of the analytical development activities is to create robust methods which are suitable for their intended purpose to support the various aspects and stages of product development. In addition, methods which are intended for GMP release and stability testing of clinical/commercial materials are expected to be validated in a phase-appropriate manner. Although such a requirement does not apply to methods used to support non-GMP process or product development activities, it is essential that the performance characteristics of these methods are evaluated and documented prior to use (this includes determining the bias relative to the proposed QC methods) since the results obtained with the inprocess methods are relied upon to make key process development decisions (Figure 2). A structured approach to method development can significantly reduce the time required to create robust methods and to establish their suitability for use.
Although a systematic development process is generally applicable, the approach described in this paper is primarily used to accelerate the development and validation of GMP methods.
The method development process for GMP methods can be divided into four distinct stages: (1) method definition, (2) technology platform selection, (3) development (including optimization with design of experiments), and (4) method performance qualification/validation. An overview of the different stages of the method development process is provided in Figure 3.
The outcome of the development process is 1) a defined analytical procedure with detailed instructions describing how the method is performed, 2) a method development report with supporting information justifying the conditions and replication strategy established for the method, 3) a method control strategy, defined based on an assessment of the major sources of variability associated with the key steps in the method, to ensure that the method performs consistently during routine use, and 4) a method qualification/ validation report documenting the phase appropriate validation of the method. The following sections describe the output for each of the four method development stages.
Analytical target profile (method definition)
The first step of the method development process is to define the method based on its intended use to monitor one or more relevant quality attributes of the NBE (i.e. scope, purpose, material type, method category, and performance expectations). The analytical target profile (ATP) serves as a prospective summary of required method characteristics (e.g. accuracy, precision) and contains the proposed target acceptance criteria which have been established for the NBE. The ATP ensures that the developed method will be suitable for its intended purpose, i.e. measurement of the quality attributes of the drug substance, drug product or in-process samples with appropriate accuracy, precision, and/or sensitivity. The analytical target profile is a living document which is reviewed and/or updated at the different stage gates of the development program to guide further method development work as knowledge of the product and the methods increases. Other reasons for updating the ATP can include a change in the method requirements resulting from regulatory feedback or changes in the manufacturing process or formulation. The key elements of the ATP are summarized in Table 1.
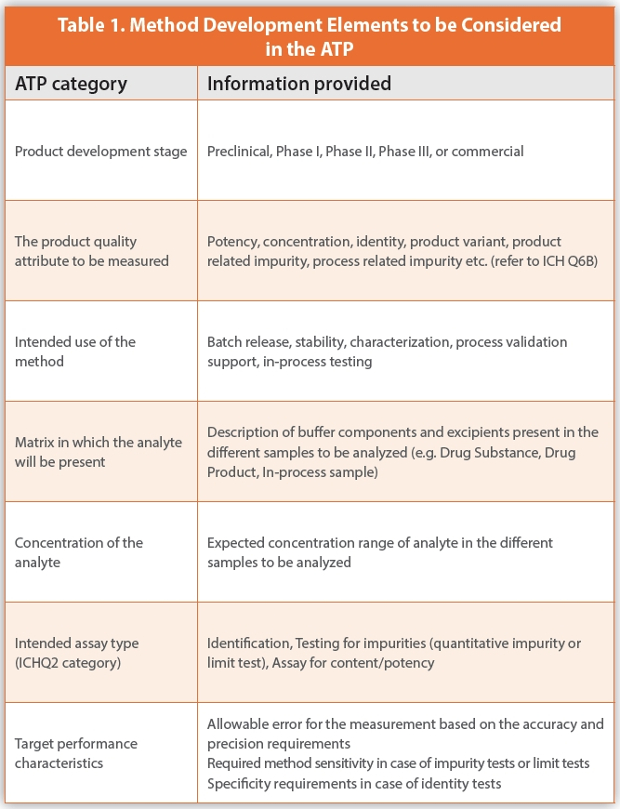
In addition to the elements listed in Table 1, the ATP should also capture specific requirements or constraints which impact the method selection or development. This may include
- Method-specific performance attributes (e.g., calibration model, technology preference)
- Business needs (e.g., duration of analysis, cycle time and throughput, capacity, required standards, controls, and reagents)
- Operational needs (e.g., lab constraints, skills or capability in the QC laboratory, safety constraints such as compliance with the REACH regulation in Europe and/or the need to minimize excessive reliance on manual pipetting to avoid repetitive motion injuries)
The target performance characteristics for the methods provided in the ATP (i.e. accuracy, intermediate precision, range, quantitation limit, detection limit, sensitivity, specificity, target measurement uncertainty etc.) should be established based on project requirements and take into consideration the applicable regulatory expectations, industry practice, and internal company standards or practices.
Technology platform selection
The starting point of technology platform selection is to gather information regarding the most suitable techniques or approaches based on the quality attribute that needs to be evaluated taking into consideration the required performance characteristics specified in the ATP. Knowledge gathering can be based on literature searches, evaluation of prior internal development work and/or through communication with research teams or other development groups within the company.
For common modalities a panel of platform methods can be established and qualified. These methods can be leveraged for new molecules within the same class by confirming their “suitability for use” through proof of concept (POC) experiments as outlined in the decision tree in Figure 4. Proof of concept experiments can be notebook driven and are designed to evaluate the method performance (e.g. accuracy, precision, sensitivity, resolution) using representative material of the new molecule in the relevant matrices (e.g. protein A eluate, DS, DP) and across the expected concentration range. The purpose of these experiments is to determine whether the new molecule is compatible with the existing platform methods by comparing the results obtained from the POC experiments against the historical experience with these methods as well as against the ATP requirements. If the method is intended to be stability-indicating, degraded material should be evaluated as part of the POC assessment to ensure relevant impurities can be detected.
Once initial compatibility with the platform method has been established, limited experiments are performed at the extremes of the proven acceptable ranges for the key product specific Analytical Quality Attributes (AQA) previously established for the method. These attributes are most often associated with the sample preparation unit operation and, based on platform risk assessments, would have been associated with high risk scores in the method assessment documents supporting the platform methods as explained in the following section. Consistent performance of the method at the extremes of the design space, when used with a new molecule, will confirm that the method remains robust and is performing as intended. However, if significant changes in the method parameters are required to obtain acceptable performance of a platform method with a new molecule, resulting in setpoints outside of the previously established design space, the platform method is considered not suitable and a new method would need to be developed.

To ensure consistency, job aids can be developed to describe the relevant POC experiments required to show that the platform method is suitable for use for a new molecule. The job aids should contain experimental strategies and designs for confirming the analytical quality attributes and provide instructions for performing a (statistical) comparison of the results against the historical performance data available for the platform methods. If the molecule fits the platform a significant reduction in method development cost and time can be realized during early stage development by leveraging the method performance data available for the platform method. In late stage, if the drug substance and drug product still fit the platform, additional savings can be realized since the robustness experiments required to verify the design space of the platform AQAs can be more limited in scope, thus saving significant resources and time.
Method development
Once the analytical technology platform for a new method has been selected (e.g. HPLC, ELISA) and the basic method parameters have been established, systematic method assessments can be used as an efficient tool to holistically evaluate and document the potential impact of the different method parameters on the ability of the method to consistently generate accurate and precise results.
An effective approach for performing method assessments relies on categorizing the individual instructions in the proposed method into different unit operations (e.g. reagent preparation, sample preparation, instrument set-up, and data analysis) as shown in Figure 5. For each of the instructions the potential failure mode is identified and the expected impact of systematic or random changes in a method parameter on the accuracy, precision, and sensitivity of the method is assessed using a three-tiered ranking system (low (1), medium (3), and high (5)). In addition, the impact is weighted based on the likelihood that the failure mode occurs (likely (1) or unlikely (5)). The expected impact of each evaluated parameter on method performance is subsequently captured as a risk probability number (RPN) which is the product of the scores assigned to the individual attributes divided by the likelihood score. The RPN is sorted in descending order to identify the factors posing the highest risk to accuracy, precision or sensitivity of the method thus allowing a prioritization of the development experiments. The method assessment is completed by late stage development and should be updated as knowledge regarding the method under development increases.
Interactions between method parameters create additional challenges during the method development process. If interactions are suspected, properly powered design of experiments (DOEs) should be performed to establish the optimal design space for the combined set of interacting parameters. In most cases the interactions between parameters are constrained within a method unit operation thus simplifying the experimental design needed to find suitable conditions to meet the ATP requirements and define the design space.
Once the method parameters have been optimized the replication strategy should be defined taking into consideration the variability of the method and the ATP requirements. Based on the established strategy the calculations to obtain the final reportable result for the method, including the number of significant figures, should be described in the analytical procedure. Only the final reportable results are compared to the acceptance criteria for the sample in case a disposition needs to be performed.

Finally, a method control strategy is established to provide assurance that the method continues to perform as intended during routine use. The method control strategy should include regulatory or compendial requirements/expectations and consider the critical method parameters as well as requirements for critical reagents, standards and/or control samples. In addition, it should define the system suitability criteria and sample acceptance criteria. The elements of the method control strategy should be captured in the analytical procedure and explained in the development report.
Method performance qualification/validation
For platform methods, which are leveraged to support early stage programs, there is limited need to collect molecule-specific qualification data. The qualification results obtained when the methods were originally established, combined with the information collected as part of the performance monitoring of the method when used routinely for other programs, provide a robust data set to show the suitability and consistency of the method. Appropriately designed and documented “fit to platform” proof of concept experiments are therefore sufficient to confirm that the methods are suitable for use.
For newly developed methods, phase appropriate qualification/validation is performed upon completion of the development activities to confirm the consistency of the method and to demonstrate that it has met the requirements of the ATP. The scope of the validation activities is defined based on the method type and the clinical phase of the program. For early stage programs the Analytical Target Profile combined with an approved standard operating procedure which describes the approaches required to evaluate the validation characteristics for different method types can be used to avoid the need to generate individual protocols for each validation exercise. However, for late stage methods, formal validation protocols and reports should be used to prospectively describe the validation activities and document the results.
Conclusions
Expedited analytical method development for therapeutics can be achieved using a well-designed strategy consisting of the following key elements:
- An analytical target profile, established based on the proposed analytical control strategy, to define the method requirements prior to initiating method development. The method requirements should be established in collaboration with other functions, in particular the process and formulation development teams.
- Systematic method assessments to consistently focus development activities on method parameters with the highest impact on method performance.
- Implementation of platform methods for common modalities across multiple projects (and multiple laboratories, if available) to minimize the need for new method development.
- Creation of method development job aids to maintain consistency when performing method assessments or fit to platform assessments. These job aids can also include definitions around platform fit considerations to facilitate platform discipline.
- Implementation of procedures for phase-appropriate method validation and creation of standardized templates for the ATP, method development reports, and validation protocols/reports to ensure consistency and to minimize writing and reviewing time.
References
- ICH Q2 – International Conference on Harmonization Q2 (R1) 2005 - Validation of Analytical Procedures: Text and Methodology
- ICH Q3A(R2) – Guidance for Industry- Impurities in New Drug Substances
- ICH Q3B(R2) –Impurities in New Drug Products
- ICH Q3C(R6) –Impurities: Guideline for Residual Solvents
- ICH Q3D(R) - Guideline for Elemental Impurities
- ICH Q5E –Comparability of Biotechnological/Biological Products
- ICH Q6B – Specifications: Test procedures and acceptance criteria for biotechnological/ biological products
- FDA: 2015 Guidance for Industry: Analytical Procedures and Method Validation for Drugs and Biologics
- USP <1220> The Analytical Procedure Lifecycle
- USP <1224> Transfer of Analytical Procedures
- PDA Technical Report 57, Analytical Method Validation and Transfer for Biotechnology Products
- PDA Technical Report No. 57-2, Analytical Method Development and Qualification for Biotechnology Product
Author Biographies
Dr. Xiaoyang Zheng is currently an Associate Director in the Analytical Development Department at Takeda. Prior to joining Takeda, she worked for 11 years at Sanofi Genzyme where she led her team to support method development/qualification, product structural characterization, control strategy establishment and corresponding CMC regulatory filings as a Principal Scientist, Analytical Team Leader and Group Leader. She was an employee of Sanofi at the time of the study. She received her Ph.D. at Northeastern University in 2007. Her doctoral research focused on proteomics method development/application in biomarker discovery and biotherapeutics development.
Scott Sacra obtained his Bachelor’s degree in Biology and Chemistry from Cornell University in 2010. He worked for four years at Teledyne Leeman Labs as the Standards Lab Chemist. Over the last 4 years at Sanofi, Mr. Sacra has worked on HPLC, UV spectroscopy and PCR method development, as well as enzyme replacement therapy (ERT) and mAb program coordination.
Marc Verhagen Ph.D. is the Senior Scientific Director Analytical Development in the Biologics Development group at Sanofi. Dr. Verhagen has been active in the biopharmaceutical industry for nearly 20 years and during this time has held positions of increasing responsibility leading analytical development activities for multiple early and late stage biologics development programs as well as commercial products. Dr. Verhagen received his B.Sc. and Ph.D. in Biochemistry from the Agricultural University in Wageningen, the Netherlands.
After obtaining her PhD from the University of London in 2001, Claire Davies performed her postdoctoral work at the William Harvey Research Institute (WHRI) at Bart’s and The London, Queen Mary’s School of Medicine and Dentistry and Joslin Diabetes Center, Harvard Medical School, Boston. Over the last 14 years, Dr. Davies has led analytical and CMC teams in product development and analytical method development and validation. Currently, Dr. Davies leads Bioanalytics, a group responsible for developing methods and strategies to support process development, product characterization and release and stability testing for therapeutic proteins and gene therapy products in preclinical and clinical development.