Introduction
It is frequently reported that the percentage of drug candidates that are limited by poor solubility is increasing [1,2]. These poorly soluble compounds typically require enabling formulations, and this trend creates challenges for teams in discovery and development who must drive in-vivo exposures high for animal toxicology studies and deliver robust dosage forms for clinical evaluation. Many enabling technologies are available for the formulator to consider, including lipids, cosolvents, surfactants, nanoparticles, cyclodextrin complexes, amorphous solid dispersions, and others. The suitability of the particular formulation approach depends largely on the physicochemical properties of the active pharmaceutical ingredient (API). Amorphous solid dispersions (ASDs) are particularly attractive for many poorly soluble drug candidates because these formulations offer many of the advantages of more conventional solid oral dosage forms but they also provide faster dissolution rates and higher drug concentrations in the gastrointestinal milieu [3]. Further, typical excipients utilized in production of ASDs are commercially available and they have proven to be well tolerated in vivo. We have successfully employed ASD technology to drive high plasma exposures in toxicology studies as well as to deliver challenging molecules in clinical studies. In this article we will discuss approaches for preparing, screening, characterizing, and dosing ASDs in preclinical and early development.
Methods of Preparation
Rotary Evaporation
Rotary evaporation is a desirable method for preparation of ASDs for early stage (pre-clinical efficacy/toxicology, Phase I) studies. This approach is fast, material sparing, relatively inexpensive, and readily available. Moreover, a wide range of batch sizes from mg to kg quantities may be prepared with high yield. ASD preparation by rotary evaporation is carried out by first dissolving the API and formulation components (polymers, surfactants) in a pharmaceutically acceptable solvent. Typical solids load in the solvent is 5% to 25% by weight, and this is generally dictated by API/polymer solubility. The solvent is then removed in a rotary evaporator using heat (typically 40 to 80 oC) and vacuum. Total time for solvent evaporation can range from minutes to hours.
Because of the relatively long evaporation time, the compound must have adequate chemical stability in the solvent at elevated temperatures. Longer evaporation times may lead to physical instability, so care must be taken to avoid API crystallization during solvent removal. This issue can be mitigated to a large extent through optimization of the temperature, vacuum, rotation, and total solids load. After removal of the solvent, the resulting ASD is isolated, dried, and milled to the desired particle size. Secondary drying in a vacuum oven or tray dryer is often employed to remove any residual solvent that remains in the final ASD powder.
Table 1 - Properties of Polymers Commonly Used in ASDs [7]

The polymers that may be employed for ASD preparation by rotary evaporation are limited to those that can be easily isolated in high yield after solvent removal (Table 1). Hydroxypropylmethyl cellulose (HPMC) based polymers are typically not amenable to rotary evaporation, as these polymers often result in a glassy film that is difficult to isolate.
While preparation of ASDs by rotary evaporation is ideal for the early stages (pre-clinical to Phase I) of drug development, it is not well suited for later stage development, manufacturing, and commercialization. The process is not readily scaleable beyond quantities on the order of 10 kg because solvent volumes become too large, leading to very long and unrealistic evaporation times. Therefore, bridging to larger scale spray drying or hot melt extrusion (HME) processes is required if the drug candidate progresses into later stage development.
Spray Drying
Spray drying is another method that is commonly utilized for the preparation of ASDs of poorly soluble compounds [4]. The method is readily scaleable from gram-sized batches during discovery and early development to kg and metric ton quantities during later stage manufacturing and commercialization. The first step in the spray drying process is to prepare a feed solution of the API and formulation components (polymers, surfactants) in a pharmaceutically acceptable solvent. The total solids load in the feed solution is typically 5% to 25% by weight, and this is generally dictated by API/polymer solubility as well as viscosity of the solution. The feed solution is then pumped into a spray nozzle along with inert, hot (typically 60 to 100 oC) drying gas where it is atomized and sprayed into a drying chamber. The solvent quickly evaporates during this process, leaving behind spray dried dispersion particles. These particles are collected in a cyclone with attached baghouse filter. Secondary drying in a vacuum oven or tray dryer is often employed to remove any residual solvent that remains in the final ASD powder. Because solvent evaporation time is extremely fast (on the order of seconds), spray drying is particularly advantageous for preparing ASDs of compounds with poor thermal stability.
There is no limitation to the types of polymers that may be employed for preparation of ASDs by spray drying. In particular, spray drying enables the preparation of ASDs in HPMC based polymers, which is often difficult, if not impossible, to achieve using rotary evaporation or HME processes. Spray drying also offers the opportunity to optimize particle size and bulk powder properties through process parameter optimization and also through the type of spray nozzle (e.g. two-fluid, ultrasonic, rotary, and pressure nozzles) [4]. In general, particle size increases with equipment scale, as a result of larger droplet sizes and longer drying residence times. This may present difficulties during development, as particle size of the spray dried powder is inherently changing as the formulation is scaled. This can be especially challenging during discovery and early development, because smaller batch sizes lead to inherently small particles (~10 μm) which can lead to issues with flow and compressibility during downstream processing. Issues with particle size can be largely mitigated via dry granulation of the spray dried powder, however this adds another relatively complicated unit operation to the overall process. Another challenge to employing spray drying during discovery and early stages of development is that the currently available lab scale spray dryers suffer from poor yield and generally cannot work on mg quantities of material.
Hot Melt Extrusion
Hot melt extrusion (HME) is the most widely used method of preparation for ASDs for commercial products, because it is particularly well suited for large scale manufacturing [5,6]. The preparation of ASDs by HME typically involves the use of twin screw extruders to mix multiple materials (API, polymer, surfactant) into a melt which is extruded through a die. The extrudate is then cooled and either shaped by calendaring or pelletized and milled to a desired particle size. The final milled extrudate is then typically blended with additional excipients and compressed. Direct shaping to a final dosage form is also possible with calendaring or injection molding technology. HME is advantageous for commercial manufacturing because it is a continuous and easily scalable process. Unlike rotary evaporation and spray-drying, HME does not require the use of organic solvents, thus it is a “green” process that reduces cost and alleviates safety/environmental concerns. Processing must be performed at temperatures above the Tg of the polymer and high enough for the API to either melt and/or dissolve into the polymer matrix. HME can be limited in the ability to process heat sensitive and/or high melting point drugs and it is generally not amenable for manufacturing small (mg to g) quantities needed in preclinical development.
Selection of Excipients
Polymers
Polymers are critical components in ASDs because they act as carriers for the drug and they inhibit crystallization in both the dosage form and in-vivo. By remaining in an amorphous state during dissolution, the drug can achieve supersaturation and potentially greater absorption, when solubility is the limiting factor.
In addition to in-vivo performance considerations, polymer properties such as the glass transition temperature (Tg), solubility in organic solvents, and hygroscopicity must be considered in order to make the ASD stable and manufacturable. The properties of some commonly used polymers for preparation of ASDs are summarized in Table 1. The polymer Tg is an important property to consider when preparing and selecting an ASD formulation. Polymers with higher Tg have less mobility, lending to better inhibition of drug crystallization. Additionally, the polymer Tg is particularly important for hot melt extrusion, as the process must be carried out above Tg to sufficiently mobilize the polymer. Organic solvent solubility of the polymer is a critical factor when manufacturing by rotary evaporation or spray drying to ensure that the polymer can be fully dissolved at the required concentration. The hygroscopicity of the polymer must also be considered, because an increase in moisture content can negatively affect physical and chemical stability, and proper packaging may be needed for ASDs composed of hygroscopic polymers.
Surfactants
Table 2 - Properties of Surfactants Commonly Used in ASDs [7]

Surfactants are often used as solubilizers or emulsifying agents in ASDs. Their primary purpose is to increase the apparent aqueous solubility and bioavailability of the drug. The properties of some common surfactants used in ASDs are listed in Table 2. As with polymers, solubility in organic solvents is an important consideration when preparing ASDs from solvent. In the case of hot melt extrusion, surfactants can have a plasticizing effect, which allows processing at lower temperatures.
Organic Solvents
Table 3 - Properties of Organic Solvents Commonly Used for Preparation of ASDs [8]
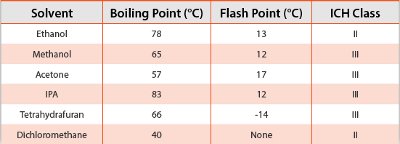
Solvents are necessary when preparing ASDs by rotary evaporation or spray drying. The properties of some common solvents used for ASD preparation are listed in Table 3. Solubility of the drug typically drives the solvent selection process, but all components should be completely dissolved to produce a homogeneous feed solution and a consistent final ASD powder. The solubility of the components in the chosen solvent must be high enough to manufacture at a reasonable throughput (typically > 5% weight of the total solids load). Water is often employed as a cosolvent for drugs (e.g. hydrates) that exhibit maximum solubility in a water-organic solvent mixture. The boiling point of the solvent is used as a guideline to set process temperatures in both rotary evaporation and spray drying processes. Sometimes a system with multiple organic solvents can be used to improve the solubility of various components. For GLP/GMP manufacturing, the ICH limit of the chosen solvent must be considered and secondary drying is often necessary to remove residual solvent.
Small-Scale ASD Screening
When conducting a screen for ASDs, the primary objective is to identify a formulation that enables in-vivo exposure of a poorly water-soluble compound and one that is also stable, both chemically and physically. For this, a wide range of polymers and polymer-surfactant systems can be screened. In addition, the drug and surfactant loading in the ASD can be evaluated for its effects on release behavior and in-vivo performance, as well as physical and chemical stability. If multiple combinations of polymers, surfactants, and drug/surfactant loads are screened, the number of samples can easily range into the hundreds.
For early stage screening, a centrifugal solvent evaporator can be used to quickly prepare a large number of samples, in parallel, using mg quantities of material. Samples can be prepared using 96-well plates for small (mg) scale screening or gram-scale quantities can be prepared using scintillation vials or small beakers [9]. Small samples allow for larger, more comprehensive screens to be carried out quickly while still providing enough material for meaningful characterization. Potential lead formulations can then be manufactured on a larger scale for further evaluation, including physical and chemical stability studies, in-vitro release characterization, and in-vitro studies in animals.
Characterization
Characterization of an ASD is a critical requirement following preparation in order to be confident in the performance of the formulation. Characterization should include analyses of both solid form and in-vitro API release in aqueous media. Numerous methods are available for characterization, the more common of which are described in the following sections and in Table 4.
Table 4 - Common Techniques Employed for ASD Solid Form Characterization

Solid Form Evaluation
Of primary importance is the identification of residual crystalline character as this signals potential long-term physical instability of the amorphous API. Polarized light microscopy (PLM) and powder X-ray powder diffraction (PXRD) are rapid, non-destructive techniques employed as an early screen for crystallinity. Individual crystals may be identified via microscopy while PXRD diffractograms provide information regarding the gross amorphous or crystalline character of the solid. Data from both methods provide greater insight when combined with more sensitive thermal methods such as differential scanning calorimetry (DSC).
DSC is employed to study transitions in the solid state as a function of temperature. Most often, ASDs are evaluated for the presence of melt endotherms and Tg. Observation of melt endotherms confirms the presence of crystalline components. Tg is an important descriptor that can provide insight into the long term stability of the ASD. Since a higher Tg generally indicates better stability, a single, high Tg value is desired for a particular ASD. Tg generally decreases with increased moisture uptake, which makes the API more prone to crystallization at higher RH conditions. Multiple Tg values indicates heterogeneity in the system and an increased potential for phase separation within the solid leading to crystallization. Thermogravimetric Analysis (TGA) is a thermal method that is complementary to DSC. TGA is employed to study weight loss from an ASD as a function of temperature and this technique is typically employed to roughly quantitate amounts of residual water or organic solvent present in the ASD. It is possible to determine the identity of materials lost during heating by coupling the TGA to a mass spectrometer (TGA-MS). These data may then be utilized to adjust processing or storage parameters to minimize the amount of plasticizing solvents which may lead to physical instability for the ASD. Dynamic vapor sorption can also be performed to understand the hygroscopic tendencies of the ASD powder.
Many other analytical methodologies are available to further characterize ASDs, although these are typically less common. Some of the other techniques include solid-state NMR spectroscopy [10], Raman spectroscopy [11], infrared spectroscopy [12], and isothermal microcalorimetry [13].
Long term stability of ASDs should be evaluated due to the increased risk of both physical and chemical instability associated with amorphous solids. Accelerated stability studies should be conducted under stressed conditions (e.g. open dish at 40°C/75% RH, 60°C/75% RH, etc.) to understand both the physical stability of the ASD and any increased risk of chemical reactivity in the presence of excipients. Photostability should also be examined to determine whether special packaging is required to prevent photochemical reactions in the amorphous state. Understanding the stability of an ASD allows one to reformulate as necessary by varying drug load, by adding antioxidants, or by selecting alternative polymers/surfactants to maximize stability.
In-vitro Release
Release of API from an ASD may be studied by a variety of in-vitro methods. One way to evaluate release from ASDs is by simple powder dissolution. This is performed by transferring a known mass of material into a known volume of biologically relevant dissolution medium (e.g. simulated gastric/intestinal fluid) under constant stirring. Solution concentrations are measured as a function of time to generate a concentration versus time release profile for the API. Solution concentrations in significant excess of the equilibrium API solubility in the particular medium should be targeted to test the ability of the ASD to achieve and maintain supersaturation.
However, it is desirable to be as biorelevant as possible to predict in-vivo performance. More dynamic techniques, such as the artificial stomach duodenum, and other multicompartmental dissolution/ release methods, should be employed to understand API release from ASDs [3,14]. These methods are used to understand complex release phenomena and to begin building in-vitro/in-vivo correlations. A dynamic dilution scheme reported by Gao et al, was recently shown to provide valuable inputs for predictions of pharmacokinetics (PK).
Dosing Considerations
Since ASDs are inherently metastable systems, it is important to consider the implications of the dosage form and dosing conditions on physical/chemical stability, manufacturability, and in-vivo performance. Aqueous suspensions enable dosing of higher ASD amounts and they are therefore ideal for dosing to animals for toxicological evaluation. Care must be taken to ensure that the ASD does not crystallize in the aqueous suspension during preparation and dosing, which could compromise in-vivo performance. Gelling and/or foaming can occur when suspending an ASD in aqueous solution. This issue may be overcome through optimization of the ASD loading in the suspension and/or addition of anti-gelling/foaming agents. When dosing smaller animals by oral gavage, the particle size of the ASD in aqueous suspension must be small enough to pass through the gavage tube.
Hard gelatin capsules (HGC) are ideal for dosing of the dry ASD to larger animals and humans. If long term stability is required, the physical and chemical compatibility of the ASD with the capsule shell must be considered, especially for hygroscopic polymers which may dry out or cause the HGC to swell depending on the RH conditions.Tablets are generally the preferred dosage form for ASD formulations of commercial products. Formulation of the ASD intermediate with secondary excipients (e.g. fillers, disintegrants, lubricants, glidants) is typically required in order to optimize the flow and compressibility of the ASD formulation to enable manufacturability of the finished tablet. Thus, the ASD intermediate must be physically and chemically compatible with the API/ ASD powder. The particle size of the ASD powder is also an important consideration for tablet formulations, as the particle size can affect the manufacturability (i.e. flow and compressibility) as well as the dissolution/release rate of the API from the tablet matrix. ASDs made by HME or rotary evaporation processes are typically milled to the optimal particle size for tableting. The desired particle size of spray dried ASDs may be obtained through process optimization, especially on large scale spray dryers. Spray dried powders made on a smaller scale during early development tend to have inherently small particle size, thus these powders may require dry granulation (roller compaction, milling) in order to obtain the desired particle size for optimal tablet properties.
Conclusions
We have outlined the fundamentals of preparing, screening, characterizing, and dosing amorphous solid dispersions. Using these methods we have successfully delivered poorly soluble drug candidates for both preclinical and clinical studies and we believe that the use of ASD technologies will continue to increase. As the science of this drug delivery approach evolves and as new excipients become available, formulators will be able to design ASD systems that are even more sophisticated and that will ultimately get new medicines to patients faster.
References
- C. J. Lipinski, Pharmacol. Toxicol. Methods, “Drug-like properties and the causes of poor solubility and poor permeability”,2000, 44, 235-249.
- E. H. Kerns, J. Pharm. Sci.,“High-throughput physicochemical profiling for drug discovery“, 2001, 90, 1838-1858.
- Gao, Y., Carr, R.A., Spence, J.K., Wang, W.W., Turner, T.M.; Lipari, J.M.; Miller, J.M., “pH-Dilution Method for Estimation of Biorelevant Drug Solubility along the Gastrointestinal Tract: Application to Physiologically Based Pharmacokinetic Modeling”, Molecular Pharmaceutics, in press.
- Dobry, D.E., Settell, D.M., Baumann, J.M., Ray, R.J., Graham, L.J., and Beyerinck, R.A., “A Model-Based Methodology for Spray-Drying Process Development”, Pharm Innov. 2009, 4, 133–142.
- Breitenbach, J.; Maegerlein, M., “Melt-extruded molecular dispersions”, Drugs and the Pharmaceutical Sciences, 2003, 133, 245-260.
- Breitenbach, J., “Melt extrusion can bring new benefits to HIV therapy: the example of Kaletra tablets”, American Journal of Drug Delivery, 2006, 4, 61-64.
- Handbook of Pharmaceutical Excipients, 6th Edition, Edited by Rowe R.C., Sheskey, P.J., and Quinn, M.E., Pharmaceutical Press, 2009.
- Miller J.M., Blackburn A.C., Macikenas D., Collman B.M., and Rodríguez-Hornedo N., “Solvent Systems for Crystallization and Polymorph Selection”, in Solvent Systems and Their Selection in Pharmaceutics and Biopharmaceutics, AAPS Biotechnology: Pharmaceutical Aspects, Volume 6, 2007.
- Shanbhag, A., Rabel, S., Nauka, E., Casadevall, G., Shivanand, P., Eichenbaum, G., Mansky, P., “Method for screening of solid dispersion formulations of low-solubility compounds-Miniaturization and automation of solvent casting and dissolution testing” International Journal of Pharmaceutics, 2008, 351, 209-218.
- Lubach, J.W., Munson, E.J., “Solid-State NMR Spectroscopy”, in Polymorphism: in the Pharmaceutical Industry, Edited by R. Hilfkiker, 2006, Wiley-VCH, 81-93.
- Breitenbach, J., Schrof, W., Neumann, J., “Confocal Raman-Spectroscopy: analytical approach to solid dipsersions and mapping of drugs“, Pharm. Res., 1999, 16, 1109-1113.
- Broman, E., Khoo, C., Taylor, L.S., “A comparison of alternative polymer excipients and processing methods for making solid dispersions of a poorly water soluble drug”, Int. J. Pharm., 2001, 222, 139-151.
- Sebhatu, T., Angberg, M., Ahlneck, C., “Assessment of the degree of disorder in crystalline solids by isothermal microcalorimetry”, Int. J. Pharm., 1994, 104, 135-144.
- Alonzo, D.E., Zhang, G.G.Z., Zhou, D., Gao, Y., Taylor, L.S., “Understanding the Behavior of Amorphous Pharmaceutical Systems during Dissolution”, Pharmaceutical Research, 2010, 27, 608-618.
Author Biographies
Brian Padden is Section Manager of Pharmaceutics at Abbott Laboratories. He holds B.A. degrees in physics and chemistry from Saint Mary’s University (Winona, MN), and M.S. and Ph.D. degrees in chemistry from the University of Minnesota. Dr. Padden started his career at the Schering-Plough Research Institute, where he was responsible for solid-state method development, GMP validation, and technology transfer to international manufacturing sites. Some of the commercial products that he contributed to during that time include Claritin®, Clarinex®, Asmanex®, Nasonex®, Noxafil®, and Zetia®. Dr. Padden then served in positions of increasing responsibility in the areas of preformulation and discovery support. In 2006, he joined Abbott and in his current role he is responsible for advancing the pipeline through material-sparing characterization and enabling preclinical formulations, in the therapeutic areas of oncology, neuroscience, pain, and dyslipidemia. He has received many technical awards, including the Abbott President’s Award in 2009, and he is a certified Lean Six Sigma Black Belt. Dr. Padden has published and presented dozens of papers, posters, and invited talks, and he is also a board member of the NSF Center for Pharmaceutical Development.
Jonathan Miller is a Principal Scientist in the Global Formulation Sciences - Solids group at Abbott Laboratories. He is also Adjunct Assistant Professor in the Department of Pharmaceutical Sciences at the University of Michigan. He has over 12 years of industrial experience both at Pfizer and now Abbott Labs, building broad expertise in the areas of pre-formulation, solid form screening/selection, formulation, biopharmaceutics, and material science. He has authored over 20 scientific publications and is an inventor on more than 10 patents and patent applications. In his current role at Abbott, he is responsible for the development of enabling formulations for poorly soluble compounds including amorphous solid dispersion formulations, lipid based drug delivery systems, and nano-particles. Dr. Miller holds a Ph.D. in Pharmaceutical Sciences and an M.S. in Pharmaceutical Engineering from the University of Michigan. He obtained his B.S. in Biochemistry from Bowling Green State University.
Timothy Robbins is the Operations Manager of the Chemical Pilot Plant at Abbott Laboratories. Dr. Robbins joined Abbott in 1993 after completing post-doctoral work at the University of California, Los Angeles. He has over 17 years of chemical research and scale-up experience. He has 12 scientific publications and is an inventor on more than 7 patents and patent applications. In his current role at Abbott, he is responsible for the running of the Chemical Pilot Plant and Kilo Lab. Prior to joining the Chemical Pilot Plant, Tim worked on a number of projects as a process research chemist within the API Process R&D organization. Dr. Robbins holds a Ph.D. in Organic Chemistry from the Unviversity of Nevada-Reno. He obtained his B.S. in Chemistry from Olivet Nazarene University.
Philip Zocharski is a Senior Scientist in the Pharmaceutics group at Abbott Laboratories. Over his 12 year career in the pharmaceutical industry, Philip has focused his research and development efforts in the areas of pre-formulation and biopharmaceutics as a colleague of Parke-Davis/Pfizer and Abbott Labs. In his current role, he applies biopharmaceutics principles and a wide array of drug delivery technologies including amorphous solid dispersions, nanoparticles, and lipids to address the specific needs of teams in early Discovery. Philip holds an M.S. in Pharmaceutical Sciences from the University of Michigan. He obtained his B.S. in Chemistry from Michigan State University.
Leena Prasad is a Scientist I in the Global Formulation Sciences – Solids group at Abbott Laboratories. She graduated in 2005 from the University of Illinois at Champaign/Urbana with a B.S. in Mechanical Engineering. She joined Abbott in July of 2007. Since joining Formulation Sciences, she has actively been involved with various projects requiring solubility enhancement technologies and has worked with the group’s solid dispersion research team.
Julie Spence is a Scientist I in the Pharmaceutics group at Abbott Laboratories. She earned her B.S. in Chemical Engineering in 2001 from the University of Michigan and her M.E. in Pharmaceutical Engineering in 2002, also from the University of Michigan. Prior to joining Abbott in 2007, Julie worked as a Scientist at the Pfizer Ann Arbor, MI labs. (2002 - 2007). Over the course of her career, Julie has gained expertise in the areas of physicochemical characterization, dermatological drug delivery, biopharmaceutics, and solid form screening/selection. Julie’s current responsibilities include developing enabling formulations for toxicology studies.
Justin Lafountaine is an Associate Scientist II in the Global Formulation Sciences – Solids group at Abbott Laboratories. He graduated in 2007 from the University of New South Wales with a B.S. in Nanotechnology. After graduation, Justin worked briefly at Pharmaform before joining the Global Formulation Sciences group in September of 2007. Since then, he has actively been involved with expanding the group’s solid dispersion capabilities and implementing Process Analytical Technology.